- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Neutral Comet Assay



Published: Vol 3, Iss 18, Sep 20, 2013 DOI: 10.21769/BioProtoc.915 Views: 38627
Reviewed by: Lin FangFanglian He
Original research article
The authors used this protocol in:

Dec 2012

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
The Comet assay (or Single Cell Gel Electrophoresis assay) is a sensitive technique to detect DNA damage at the level of an individual cell. This technique is based on micro-electrophoresis of cells DNA content. Briefly, cells are embedded in agarose, lysed and submitted to an electric field, before the staining step with a fluorescent DNA binding dye. Damaged DNA (charged DNA) migrates in this field, forming the tail of a “comet”, while undamaged DNA remained in the head of the “comet”. The following document describes the protocol to realize a neutral comet assay. This assay can be applied to different cell types and has been useful for numerous applications in fields of toxicology or DNA damage and repair.
Keywords: Comet assayMaterials and Reagents
- Cells to analyze
- Low Melting Point (LMP) Agarose (Sigma-Aldrich, catalog number: A9414 )
- Seakem® Agarose (Ozyme, catalog number: LON50004 )
- PBS (Ca2+ and Mg2+-free phosphate-buffered saline)
- 5 N NaOH
- 0.5 M EDTA disodium salt solution (pH 8)
- Trisma base
- Triton X-100
- N-Lauroylsarcosine (Sigma-Aldrich, catalog number: L5125 )
- Dimethylsulphoxide (DMSO)
- Absolute ethanol
- Ethidium bromide (10 mg/ml)
- Trypsin/EDTA
- Sodium acetate
- Lysis solution (see Recipes)
- Electrophoresis solution (see Recipes)
Equipment
- Microscope Super Frost plus glass slides
- Malassez chamber
- Microscope coverslips (22 x 22 mm)
- Microscope coverslips (24 x 32 mm)
- Centrifuges
- Electrophoresis tank: Econo-Submarine (20 cm x 30 cm) (C.B.S. Scientific, USA)
- Fluorescence microscope, camera and software (e.g. Nikon Eclipse 50i microscope equipped with a Luca S camera and Komet 6.0 software)
Software
- Komet 6.0 software (Andor Technology)
Procedure
- Prepare agarose solution and slides.
- At least 24 h before the experiments:
- Prepare 0.8% solution of Seakem® Agarose in PBS.
- Pre-coat Super Frost slides by dipping in a vertical jar containing melted agarose, stirred with a magnetic stirrer and kept at 100 °C (Figure 1).

Figure 1. Coating of the slides with agarose - Drain off the agarose in excess by wiping the back of the slides (Figure 2).
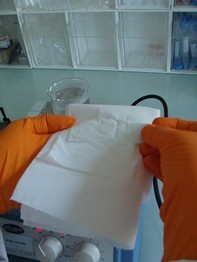

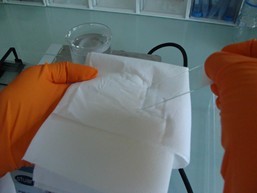
Figure 2. Slides coating with agarose. Removing of the agarose on the back of the slides. - Let the slides dry and then store at room temperature until use.
- Prepare 0.8% solution of Seakem® Agarose in PBS.
- At least 2 h before the experiments: prepare 0.7% solution of LMP agarose in PBS and place it at 37 °C in a water-bath until use.
- At least 24 h before the experiments:
- Prepare cells
The number of harvested cells can be adjusted according to the size of the cells. Cells could be numbered either by an automated cell counter or a counting chamber (e.g. Malassez chamber).
If cells used are adherent, cells must be carefully detached with trypsin/EDTA and isolated before centrifugation and further use. - Embed cells in LMP agarose (in a dark room):
- After cell centrifugation, discard the supernatant and resuspend the pellet of cells (150,000 to 200,000 cells) by gently pipetting with 200 μl of 0.7% LMP agarose.
- Lay 65 μl of agarose containing the cells on each pre-coated glass slide.
- Immediately cover with a 24 x 32 mm coverslip.
- Put the slide on an ice-pack for solidification during 5-10 minutes.
- Slide off the coverslip to remove it.
- Finally cover with 80 μl of LMP agarose (top agarose layer) and cover again with a 24 x 32 mm coverslip.
- Put the slide again on an ice-pack for solidification during 5-10 minutes.
- Remove the coverslip.
- After cell centrifugation, discard the supernatant and resuspend the pellet of cells (150,000 to 200,000 cells) by gently pipetting with 200 μl of 0.7% LMP agarose.
- Lysis and electrophoresis (in a dark room):
- Place the slides in lysis solution for at least 1 h at 4 °C.
- Wash three times for 5 min with the electrophoresis buffer.
- Transfer the slides in the electrophoresis tank filled with electrophoresis solution (Figure 3).

Figure 3. Electrophoresis of the slides. Disposition of the slides in the tank. In this case eight slides are subjected to electrophoresis, four by row. - Proceed to electrophoresis at 18 V (0.5 V/cm) during 1 h.
- Wash in PBS for 2 x 5 min.
- Place the slides in lysis solution for at least 1 h at 4 °C.
- Dehydration, staining and analysis:
- Fix the cells with 2 x 10 min washes in absolute ethanol, air-dry for at least 2 hours at room temperature.
- Add 50 μl ethidium bromide (2 μg/ml in water) on the microscope slide and cover with a 22 x 22 mm coverslip for staining.
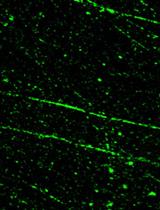
- Analyze the cells: score 50 cells per slide, 2 slides per condition with the fluorescence microscope equipped with a camera and adapted software (Figures 4 and 5).
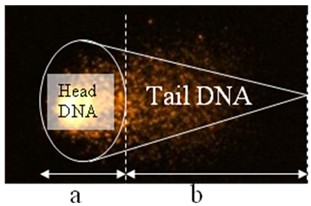
Figure 4. Representing analysis of each cell
a = head length
b = tail length
% tail DNA = fraction of DNA in the tail
Tail moment = % tail DNA x b
Comet tail length can be calculated by different ways depending on the authors. Tail moment is a common parameter used to characterize the comet. For this, the fraction of DNA in the tail is evaluated by the fluorescence in the tail and divided by the total fluorescence (in the head and in the tail) to be expressed in percentage. Tail moment is the product of % tail DNA and tail length.
Figure 5. Images representing nucleus of undamaged cells, negative for the presence of comet (left panel) and nucleus of damaged cells presenting comet (right panel)
As a positive control, cells irradiated with ionizing radiations at 20 Gy and examined just after irradiation are a good control as shown in the photographs above.
- Fix the cells with 2 x 10 min washes in absolute ethanol, air-dry for at least 2 hours at room temperature.
Recipes
- Lysis solution
2.5 M NaCl
0.1 M EDTA
10 mM Trizma base (pH 10)
1% N-laurylsarcosine
0.5% Triton X-100
10% DMSO final
Keep at 4 °C - Electrophoresis solution
300 mM sodium acetate
100 mM Tris-HCl (pH 8.3) at 4 °C
Acknowledgments
This protocol was adapted from previously published papers (Courilleau et al., 2012; Olive et al., 1990; Ostling and Johanson, 1984; Ostling and Johanson, 1987; Wojewodzka et al., 2002).
References
- Courilleau, C., Chailleux, C., Jauneau, A., Grimal, F., Briois, S., Boutet-Robinet, E., Boudsocq, F., Trouche, D. and Canitrot, Y. (2012). The chromatin remodeler p400 ATPase facilitates Rad51-mediated repair of DNA double-strand breaks. J Cell Biol 199(7): 1067-1081.
- Olive, D. M., Johny, M. and Sethi, S. K. (1990). Use of an alkaline phosphatase-labeled synthetic oligonucleotide probe for detection of Campylobacter jejuni and Campylobacter coli. J Clin Microbiol 28(7): 1565-1569.
- Ostling, O. and Johanson, K. J. (1984). Microelectrophoretic study of radiation-induced DNA damages in individual mammalian cells. Biochem Biophys Res Commun 123(1): 291-298.
- Ostling, O. and Johanson, K. J. (1987). Bleomycin, in contrast to gamma irradiation, induces extreme variation of DNA strand breakage from cell to cell. Int J Radiat Biol Relat Stud Phys Chem Med 52(5): 683-691.
- Wojewodzka, M., Buraczewska, I. and Kruszewski, M. (2002). A modified neutral comet assay: elimination of lysis at high temperature and validation of the assay with anti-single-stranded DNA antibody. Mutat Res 518(1): 9-20.
Article Information
Copyright
© 2013 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Boutet-Robinet, E., Trouche, D. and Canitrot, Y. (2013). Neutral Comet Assay. Bio-protocol 3(18): e915. DOI: 10.21769/BioProtoc.915.
- Courilleau, C., Chailleux, C., Jauneau, A., Grimal, F., Briois, S., Boutet-Robinet, E., Boudsocq, F., Trouche, D. and Canitrot, Y. (2012). The chromatin remodeler p400 ATPase facilitates Rad51-mediated repair of DNA double-strand breaks. J Cell Biol 199(7): 1067-1081.
Category
Cancer Biology > General technique > Biochemical assays > DNA structure and alterations
Cancer Biology > Genome instability & mutation > Biochemical assays > DNA structure and alterations
Cell Biology > Single cell analysis > Cell counter
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.