- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

DNase I Footprinting to Identify Protein Binding Sites
Published: Vol 3, Iss 14, Jul 20, 2013 DOI: 10.21769/BioProtoc.824 Views: 24554
Reviewed by: Fanglian He
Original research article
The authors used this protocol in:

Sep 2012
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
DNase I footprinting is used to precisely localise the position that a DNA binding protein, e.g. a transcription factor, binds to a DNA fragment. A DNA fragment of a few hundred bp is labelled at one end and then incubated with the proteins suspected to bind. After a limited digestion with DNase I, the reaction is quenched, DNA is precipitated and analysed on a denaturing polyacrylamide gel. This protocol uses 32P-radioactively labeled DNA.
Materials and Reagents
- Oligonucleotides (usually 20-30 mer) to amplify a suitable fragment (100-400 bp) encompassing the region to be tested for protein binding ability
- Plasmid DNA carrying the cloned required region to use as template for the PCR amplification
- [γ-32P] ATP (3,000 Ci/mmole, 30 μCi = 3 μl/labeling ) (e.g. NEN, catalog number: BLU502A)
- Polynucleotide kinase (PNK) (e.g. Biolabs, catalog number: M0201 )
- Agarose
- Purified protein (or enriched crude bacterial extracts see below)
- DNase I (e.g. Sigma-Aldrich, catalog number: D5025 )
- Phenol
- Chloroform
- Herring sperm DNA (e.g. Sigma-Aldrich, catalog number: D6898 ; Roche, catalog number: 223 646 )
- BSA (e.g. Biolabs, catalog number: B9001 )
- Acrylamide
- Urea
- DTTP (e.g. Biolabs, catalog number: N0447 )
- Taq polymerase (5 units/μl) (e.g. Biolabs, catalog number: M0267 )
- DNA Marker (e.g. 100 bp ladder, Biolabs, catalog number: N3231 )
- Antarctic alkaline phosphatase (Biolabs, catalog number: M0296 )
- MspI (Biolabs, catalog number: N3032 )
- 40% Acrylamide stock (19:1 acrylamide: bis acrylamide) (e.g. Euromedex, catalog number: EU0076-C)
- TBE buffer
- Binding buffer (see Recipes)
- DNase I dilution buffer (see Recipes)
- DNase I stop buffer (see Recipes)
- DNase I stock (see Recipes)
- Loading formamide dyes (see Recipes)
- Denaturing Sequencing gel (6% acrylamide) (see Recipes)
- Hepes-Glutamate (see Recipes)
Equipment
- Suitable space for working with 32P radioactivity
- Image quantification apparatus (e.g. Typhoon GE Healthcare Life Sciences; X-ray film and developing materials)
- PCR machine
- Small horizontal agarose gel apparatus
- Transilluminator (preferably 365 nM)
- Apparatus for running a 30 cm sequencing gel (e.g. Model S2 Vertical sequencing apparatus, now sold by Biometra)
- Power supply capable of producing 2,000 volts and 60 watts)
- Geiger counter to monitor for radioactivity and any contamination.
- Heating block at 90 °C
- Gel drying apparatus
Procedure
- Preparation of the labeled DNA fragment
- Label the 5' end of one of the oligonucleotides to be used to make the fragment to footprint. Choose the oligo so that the suspected binding site is not too far from the labeled end. Normally the footprint should be performed on both strands of the DNA, i.e. using two DNA fragments labeled at either end. Use a 0.5 ml tube suitable for PCR.
3 μl Oligo 1, 10 pmoles/μl
2 μl 10x PNK buffer (supplied by manufacturer of PNK)
3 μl [γ- 32P] ATP (3,000 Ci/mmole)
11 μl H2O
1 μl (10 units) PNK
Incubate 30 min 37 °C
N.B. Take suitable precautions for use of radioactivity. Perform in approved location. - Precipitate the labeled oligo. Add
80 μl 0.1 M Sodium acetate (natural pH about 9.0)
1 μl 10 mM Sodium phosphate buffer, pH 7.4
250 μl ethanol (96%)
Incubate in dry ice for 30 min (or at -80 °C for > 1 h). - Centrifuge 15 min 4 °C 12,000 x g.
- Carefully remove the supernatant.
N.B. Very radioactive, Discard accordingly. - Rinse the (tiny) pellet with 100 μl 96% ethanol (or 70% ethanol at -20 °C). Centrifuge 5 min 4 °C 12,000 x g.
- Carefully remove the supernatant. Dry in vacuo 5 min.
- Resuspend the labeled Oligo 1 in 35 μl H2O. Vortex well and give a quick centrifugation to place all labeled oligo in bottom of tube.
- Add 5 μl Thermopol buffer (Biolabs or other suitable Taq polymerase buffer).
5 μl deoxyNTP mix containing 2.5 mM each dATP, dCTP, dGTP, DTTP.
4 μl Oligo 2 10 pmoles/μl (Oligo 2 corresponds to the other end of fragment to be amplified).
1 μl template DNA (e.g. plasmid DNA carrying the cloned region to be amplified, about 50 ng depending on size of plasmid. We usually use 1 μl 1/10 dilution of standard mini plasmid DNA preparation).
Mix well, quick centrifugation and add 0.5 μl (2.5 units) Taq polymerase (5 units/μl) and immediately start PCR. - PCR cycling
- Denature 94 °C for 2 min
- 94 °C for 30 sec
- 55 °C* for 30 sec
- 72 °C for 30 sec*
- Repeat b-d 25 times
- Final extension 5 min 72 °C
- Denature 94 °C for 2 min
- Test 1-2 μl on small agarose gel (containing ethidium bromide or other suitable DNA detection reagent) for amplification of a fragment of the correct size and check you have a fragment of the correct size in good yield (i.e. most or all of the oligos have been used up) 30 pmoles of oligos can give maximally 2 μg of a fragment of 100 bp and 10 μg of a fragment 500 bp. Proceed to purification. In theory you can use the PCR as it is or after passage through a spin column. However any minor, shorter length contaminants will produce artifactual bands in the footprint and the presence of unincorporated radioactivity will not allow you to estimate the amount of radioactive DNA in the footprinting reactions. So we always proceed to purify the labeled DNA from an agarose gel. In exceptional cases e.g. to eliminate a close running contaminant band from a short (100-200 bp) fragment, the fragments can be purified on a native acrylamide gel (see step I-14 below).
- Run the PCR mixture, mixed with 10 μl of loading dyes, on a small 1% agarose gel in 50 mM TBE buffer. Usually the whole 50 μl PCR can be loaded in 3 wells.
- Visualize the gel on a long wavelength (365 nm) transilluminator (to minimize damage to the DNA by short wavelength) and cut out the agarose containing the radioactive fragment. Discard rest of gel as radioactive waste.
- Extract the DNA from agarose using a gel purification kit (e.g. Machery-Nagel Nucleo-spin Gel and PCR clean-up). Elute in 50 μl elution buffer.
- To purify the DNA from an acrylamide gel: Run the PCR mix on a native acrylamide gel (5-8% depending upon size in 50 mM TBE at room temperature i.e. do not allow the gel to heat up. Depending upon the size of the apparatus used 100-200 volts should be adequate). Locate the radioactive DNA by short exposure of the wet gel wrapped in Saran wrap to a phosphorimager screen (or X-ray film). The piece of acrylamide containing the radioactive band is cut out and the radioactive DNA eluted by shaking overnight at 37 °C in 1 ml of 0.5 M ammonium acetate, 0.1% SDS, 1 mM EDTA. Separate the aqueous phase from the acrylamide gel piece by centrifugation and transfer to another tube. Extract with 0.5 ml phenol/CHCl3 and precipitate the DNA with 2.5 volumes ethanol in dry ice for at least 30 min. Centrifuge 10 min 4 °C, remove all the supernatant, dry in vacuo 2-3 min) Resuspend in 50 μl elution buffer (as for DNA eluted from agarose.).
- Run 1 μl on a new small agarose gel and estimate the quantity by comparison to the staining intensity of marker DNAs (e.g. 100 bp ladder).
- Count 1 μl by Kerenkov radiation in a scintillation counter or estimate using a Geiger counter. Expect to have 50,000-150,000 cpm/μl with about 5-50 ng DNA/μl (yield in range 10-50% of the moles of starting oligonucleotide). You can calculate the molar concentration of the DNA fragment from the length of the fragment in bp and using 1 bp corresponds to a molecular mass of 660.
- Label the 5' end of one of the oligonucleotides to be used to make the fragment to footprint. Choose the oligo so that the suspected binding site is not too far from the labeled end. Normally the footprint should be performed on both strands of the DNA, i.e. using two DNA fragments labeled at either end. Use a 0.5 ml tube suitable for PCR.
- The footprinting reaction
- All protein and DNA dilutions are made in 1x binding buffer. Protein dilutions are made at 4 °C to minimize any instability of the protein in dilute solutions. Binding reactions can be made at RT or at 30 °C or 37 °C depending upon the experiment. E.g. Most prokaryotic transcription factors bind DNA at RT. E.coli RNA polymerase will only form open complexes at 37 °C.
- The binding buffer we usually use is Hepes-Glutamate. The concentration of K glutamate can be increased or decreased according to the experiment; generally higher salt favors specificity but decreases affinity. Mg++ salts (1-10 mM) can be added according to the experiment, e.g. Mg++ is required for RNA polymerase binding. The BSA is added to stabilize dilute proteins and prevent non-specific absorption to the microfuge tube. The reaction mix in general consists of 20 μl DNA solution to which we add 20 μl of the diluted protein (if several components need to be tested at the same time the volume for each component can be reduced accordingly to give a final volume of 40 μl).
- To test a range of protein concentrations for binding to the labeled DNA. Make a suitable volume of binding buffer and keep on ice. Prepare a series of dilution tubes for the protein to test over a range of concentrations. This range depends entirely on the protein under study as binding constants can vary from nM to nearly mM. E.g. for 10 serial dilution of 1/2 concentration each step, Prepare 10 tubes with 25 μl binding buffer on ice.
- Prepare a suitable volume of labeled DNA e.g. For 12 reactions prepare 240 μl binding buffer and add labeled DNA fragment to have about 20,000 -100,000 cpm/reaction. Mix well. Ideally this should give a final concentration of the DNA of about 1 nM in the 40 μl footprinting reaction, if binding constants of protein to DNA are in the nM range.
- Dispense 20 μl of the DNA mix into 12 1.5 ml microtubes at RT.
- Make the protein dilutions on ice and immediately mix the diluted protein with the DNA at RT. E.g. Dilute the protein to 1 μM in 50 μl binding buffer.
- Add 20 μl to one tube of DNA (to give final concentration of 500 nM). Mix by pipetting. Leave at RT until finished all mixes.
- Take 25 μl of the 1 μM dilution and mix with 25 μl binding buffer in next dilution tube.
- Add 20 μl of this dilution to the next DNA tube (final concentration 250 nM).
- Take 25 μl of the 0.5 μM dilution and mix with 25 μl binding buffer in next dilution tube.
- Continue for the whole series of 10 dilutions.
- Add 20 μl to one tube of DNA (to give final concentration of 500 nM). Mix by pipetting. Leave at RT until finished all mixes.
- Add 20 μl 1x binding buffer to two DNA samples for the (essential) controls without protein.
- Incubate for 10 min at RT (or at chosen temperature for chosen time).
- Meanwhile prepare a suitable dilution of DNase I in DNase I dilution buffer. It is crucial to find the correct DNase I concentration to get limited DNase attack so that not all the DNA is degraded. As there should in theory be no more than 1 break/DNA strand, the majority (80-90%) of the DNA should be full length.
It is usually necessary to make a pilot experiment with each new stock of DNase I to test a series of dilutions of DNase I with a DNA fragment (in the absence of binding proteins) to find the concentration which gives an adequate ladder but leaves full length DNA at the top of the gel. The amount of DNase I used should be lower for longer DNA fragments. The amount given below is a guideline only. - DNase I stock of 5 mg/ml in DNase I storage buffer (store in aliquots at -20 °C).
Dilute about 10,000 fold (e.g. 2 μl to 200 μl, in DNase I dilution buffer, then take 2 μl of the first dilution to 200 μl DNase I dilution buffer) to give 0.5 μg/ml. - To make the footprint reactions:
T= 0 add 4 μl dilute DNase I to the footprint mix 1. Mix gently by pipetting.
T= 15 sec Add 4 μl dilute DNase I to the footprint mix 2. Mix gently by pipetting.
T= 30 sec Add 4 μl dilute DNase I to the footprint mix 3. Mix gently by pipetting.
T= 45 sec Add 4 μl dilute DNase I to the footprint mix 4. Mix gently by pipetting.
T= 60 sec Add 100 μl phenol pH 8.0 to the footprint mix 1. Vortex to stop reaction
T= 75 sec Add 100 μl phenol pH 8.0 to the footprint mix 2. Vortex to stop reaction.
T= 90 sec Add 100 μl phenol pH 8.0 to the footprint mix 3. Vortex to stop reaction.
T= 105 sec Add 100 μl phenol pH 8.0 to the footprint mix 4. Vortex to stop reaction.
Repeat until all the tubes have been treated.
Precise timings are crucial to get precise and comparable DNase I digestions in each lane (some researchers prefer to use longer times and more dilute DNase I). - Add 200 μl DNase I STOP solution to each reaction. Vortex well.
- Centrifuge 10 min RT.
- Transfer the aqueous upper phase (careful not to take any phenol) to a clean 1.5 ml microfuge tube.
- Add 600 μl 96% ethanol. Mix well and incubate in dry ice for 1 h (or overnight at -80 °C).
- Centrifuge 15 min, 4 °C, 12,000 x g.
- Remove the supernatant carefully with a pipette and 1 ml pipette tip. Depending upon the number of cpm used per reaction and the sensitivity of the available Geiger counter, verify that some radioactivity is in the (invisible) pellet with a Geiger counter. If the herring sperm DNA, used in the DNase I STOP to aid precipitation, has not been sufficiently sonicated or too much has been used, the DNA pellet might not adhere to the microfuge tube and can be lost with the ethanol.
- Centrifuge again, 5 min 4 °C. Remove the rest of the liquid with a 20 μl pipette tip. Verify that the radioactivity is still in the tube.
- Dry in vacuo 5 min.
- Resuspend in 5 μl H2O and add 6 μl gel loading formamide dyes (deionized formamide with Bromophenol blue and Xylene cyanol). Vortex well. Quick centrifuge to put all liquid in bottom of tube.
- Heat to 90 °C for 2 min. Quench in ice and immediately load (5 μl) onto a denaturing (7 M urea) acrylamide sequencing gel, acrylamide (19:1 acrylamide: bis-acrylamide), which has been prerun for 30 min to 1 h to get hot. Voltage and wattage depends upon the apparatus used. We use 60 watts for the Model S2. Use with 5-10% final concentration acrylamide depending on the size of the fragment under investigation.
- To locate the site of protein binding it is necessary to calibrate gels with molecular weight markers. E.g. DNA marker pBR322 digested with MspI, treated with alkaline phosphatase (e.g. Antarctic alkaline phosphatase) and labeled with [γ-32P] ATP and PNK or a sequencing ladder prepared using the same Oligo 1, which was used for the radioactive labeling of the fragment.
- Electrophorese at 60 watts for a 30 x 30 plates and 1 mM thick gels using the Model S2 apparatus. Adjust volts and power for other sized gels. Time of migration depends upon the size of the fragment and expected position of protein binding sites (generally 80 min to 3 h).
- After migration, allow to cool slightly, open plates, transfer the gel to Whatman 3 mm paper. Cover with Saran wrap and dry on a gel drying apparatus.
- Put the dried gel, still covered with Saran to expose in a Phosphorimager cassette overnight (alternatively expose to X-ray film with an intensifying screen).
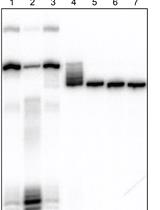
- The pattern of DNase I cleavages with and without protein indicates the site of protein binding (Figure 1). Deformations such as bent or looped DNA are indicated by hypersensitive cleavages compared to the control (free DNA), since bending of the DNA has facilitated attack by DNase I in the wider minor groove formed on the outside of the loop or bend (Figure 2).
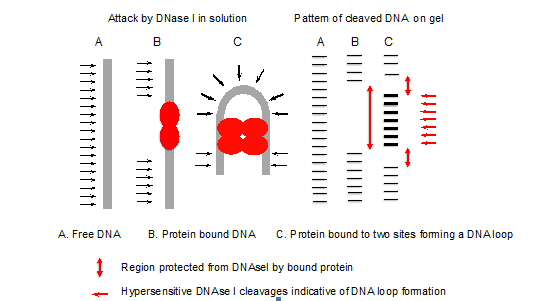
Figure 1. Principle of DNase I footprinting
Figure 2. Example of DNase I footprinting - Using the Image Quant program, it is possible to quantify the amount of radioactivity in specific bands or regions, corresponding to protein protected sites and non-protected regions, and hence to derive binding constants for the proteins.
- All protein and DNA dilutions are made in 1x binding buffer. Protein dilutions are made at 4 °C to minimize any instability of the protein in dilute solutions. Binding reactions can be made at RT or at 30 °C or 37 °C depending upon the experiment. E.g. Most prokaryotic transcription factors bind DNA at RT. E.coli RNA polymerase will only form open complexes at 37 °C.
Recipes
- Binding buffer
25 mM Hepes (pH 8.0)
100 mM K glutamate (pH 8.0)
0.5 mg/ml BSA - DNase I dilution buffer
10 mM Tris (pH 8.0)
10 mM MgCl2
10 mM CaCl2
125 mM KCl
0.1 mM DTT - DNase I stop buffer
0.5 M Na acetate pH 5.0
10 μg/ml DNA (e.g. sonicated herring sperm DNA. Suspend DNA in 10 mg ml-1 H2O and allow to hydrate overnight at 4 °C. Sonicate until the solution loses all viscosity.)
2.5 mM EDTA - DNase I stock e.g. 5 mg/ml in
50% glycerol
100 mM NaCl
10 mM Tris (pH 8.0)
10 mM MgCl2 - Loading formamide dyes
1 ml deionized formamide
10 μl 5% solution xylene cyanol and bromophenol blue. - Denaturing Sequencing gel (6% acrylamide)
6% acrylamide (19:1 acrylamide: bis acrylamide) (dilute from 40% stock)
7 M urea
1x TBE
For 80 ml of acrylamide/urea/TBE mixture add 0.4 ml 10% ammonium persulphate and 40 μl TEMED to polymerise the acrylamide, mix gently and carefully pour between the gel plates (0.4 mM thick spacers) avoiding bubbles. Insert a comb to make the wells and leave to polymerise horizontally. - Hepes-Glutamate
25 mM Hepes (pH 8.0)
100 mM K glutamatew
0.5 mg/ml BSA
Acknowledgments
The use of DNAse I to identify protein bindinging sites on DNA was first described by Galas and Schmitz (1978). Since then it has been exploited and adapted in very many laboratories. Our protocol is based on that in use in the laboratory of Annie Kolb (Colland et al., 2000; Marschall et al., 1998), who introduced the use of potassium glutamate in the binding buffer, which mimics in vivo conditions and increases the affinity of most proteins for their DNA targets. We are extremely grateful to Annie Kolb for her continuous advice and interest. Work in our lab has been funded by the Centre National de Recherche Scientifique (CNRS) to UPR9073 (now renamed FRE3630), by Université Paris 7, Denis Diderot; Agence National de Recherche [(ANR-09-Blanc 0399 (GRONAG)] and by the "Initiative d'Excellence" program from the French state [ANR-11-LBX-0011-01 (DYNAMO)].
References
- Brenowitz, M., Senear, D. F., Shea, M. A. and Ackers, G. K. (1986). Quantitative DNase footprint titration: a method for studying protein-DNA interactions. Methods Enzymol 130: 132-181.
- Brechemier-Baey, D., Dominguez-Ramirez, L. and Plumbridge, J. (2012). The linker sequence, joining the DNA-binding domain of the homologous transcription factors, Mlc and NagC, to the rest of the protein, determines the specificity of their DNA target recognition in Escherichia coli. Mol Microbiol 85(5): 1007-1019.
- Colland, F., Barth, M., Hengge-Aronis, R. and Kolb, A. (2000). Sigma factor selectivity of Escherichia coli RNA polymerase: role for CRP, IHF and lrp transcription factors. EMBO J 19(12): 3028-3037.
- El Qaidi, S. and Plumbridge, J. (2008). Switching control of expression of ptsG from the Mlc regulon to the NagC regulon. J Bacteriol 190(13): 4677-4686.
- Galas, D. J. and Schmitz, A. (1978). DNase footprinting: a simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res 5(9): 3157-3170.
- Marschall, C., Labrousse, V., Kreimer, M., Weichart, D., Kolb, A. and Hengge-Aronis, R. (1998). Molecular analysis of the regulation of csiD, a carbon starvation-inducible gene in Escherichia coli that is exclusively dependent on sigma s and requires activation by cAMP-CRP. J Mol Biol 276(2): 339-353.
- Plumbridge, J. and Kolb, A. (1991). CAP and Nag repressor binding to the regulatory regions of the nagE-B and manX genes of Escherichia coli. J Mol Biol 217(4): 661-679.
Article Information
Copyright
© 2013 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Gaugué, I., Bréchemier-Baey, D. and Plumbridge, J. (2013). DNase I Footprinting to Identify Protein Binding Sites. Bio-protocol 3(14): e824. DOI: 10.21769/BioProtoc.824.
Category
Molecular Biology > DNA > DNA-protein interaction
Molecular Biology > DNA > DNA labeling
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.