- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Cell-free Fluorescent Intra-Golgi Retrograde Vesicle Trafficking Assay
Published: Vol 7, Iss 22, Nov 20, 2017 DOI: 10.21769/BioProtoc.2616 Views: 8748
Reviewed by: Jia LiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Intra-Golgi retrograde vesicle transport is used to traffic and sort resident Golgi enzymes to their appropriate cisternal locations. An assay was established to investigate the molecular details of vesicle targeting in a cell-free system. Stable cell lines were generated in which the trans-Golgi enzyme galactosyltransferase (GalT) was tagged with either CFP or YFP. Given that GalT is recycled to the cisterna where it is located at steady state, GalT-containing vesicles target GalT-containing cisternal membranes. Golgi membranes were therefore isolated from GalT-CFP expressing cells, while vesicles were prepared from GalT-YFP expressing ones. Incubating CFP-labelled Golgi with YFP-labelled vesicles in the presence of cytosol and an energy regeneration mixture at 37 °C produced a significant increase in CFP-YFP co-localization upon fluorescent imaging of the mixture compared to incubation on ice. The assay was validated to require energy, proteins and physiologically important trafficking components such as Rab GTPases and the conserved oligomeric Golgi tethering complex. This assay is useful for the investigation of both physiological and pathological changes that affect the Golgi trafficking machinery, in particular, vesicle tethering.
Keywords: Golgi apparatusBackground
The molecular mechanisms of intracellular vesicle targeting are important to decipher to understand processes as diverse as glycosylation homeostasis, neurotransmitter release, regulation of signaling receptors and nutrient uptake (Ungar and Hughson, 2003; Fisher and Ungar, 2016). The Golgi apparatus is an excellent test case, as it maintains a network of target compartments, called cisternae, that require the specific delivery of different vesicles (Cottam and Ungar, 2012). The Golgi can also be isolated in a functional form retaining its ability for vesicle transport (Balch et al., 1984). Fluorescent labelling of vesicles and target cisternae offers a direct readout of vesicle targeting by measuring the co-localization of the two membrane fractions following a cell-free incubation. This type of measurement has some caveats. The size of vesicles is below the resolution limit of conventional microscopy, and there are only single fluorophores in the majority of the vesicles (C. Baumann and D. Ungar, University of York, unpublished data). This means that very high quality optics and sensitive detection has to be combined with automated exposure control during microscopy to avoid photobleaching, and sophisticated image processing to obtain images that are free of noise.
The assay was set up to investigate the molecular requirements of vesicle tethering at the trans-Golgi (Cottam et al., 2014). Accordingly, it was found to be dependent on functional Rab GTPases (Rabs), as the protein Rab-GDI, which extracts Rabs from membranes (Soldati et al., 1993), was found to inhibit the signal (Cottam et al., 2014). Moreover, the assay was sensitive to various defects of the conserved oligomeric Golgi (COG) tethering complex. Cytosol is an essential component of the assay mixture for obtaining activity, and when used from cells harboring patient-derived COG mutations (Wu et al., 2004; Luebbehusen et al., 2010), it inhibited the assay signal (Cottam et al., 2014). Moreover, the assay was able to differentiate the contributions to retrograde trafficking of two different COG mutants (Cog1- and Cog2-null mutations, Cottam et al., 2014), which have essentially identical cellular phenotypes in CHO cells (Kingsley et al., 1986).
Materials and Reagents
- Microscope slides and coverslips (purchased from Thermo Fisher Scientific, Thermo ScientificTM, catalog numbers: MNJ-400-030Y and MNJ-100-030J )
Note: The quality of the slides and coverslips is critical, the mentioned product does work, if others are to be used it is advised to test these. - Silica beads (5 micron silica beads) (Bangs Laboratories, catalog number: SS06N )
- 0.5 ml and 1.5 ml microfuge tubes, 0.2 ml PCR tubes
- T175 flasks (each 175 cm2) (Corning BV Life Sciences, Amsterdam, the Netherlands)
- 10 ml serological pipette
- PD-10 desalting column (GE Healthcare, Buckinghamshire, UK)
- Wild type HEK293 cells
- HEK293 cells stably expressing CFP/YFP-GalT
- Water
Note: Molecular biology grade water was used for all procedures, including the washing of microscope slides and coverslips. - Detergent decon90 (Decon Laboratories Limited, Sussex, UK)
- Potassium chloride (KCl)
- Geneticin (GibcoTM)
- Dulbecco’s Modified Eagle Medium (DMEM, high glucose, pyruvate, no glutamine) (Thermo Fisher Scientific, GibcoTM, catalog number: 21969 )
- Fetal bovine serum (FBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 12657029 )
- GlutaMAX-I (an L-alanyl-L-glutamine dipeptide substitute for L-glutamine) (Thermo Fisher Scientific, GibcoTM, catalog number: A12860-01 )
- Penicillin-streptomycin (10,000 U/ml; 100x) (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 )
- Ham’s F-12 Nutrient Mix (Sigma-Aldrich, catalog number: N4888 )
- Liquid nitrogen
- Tris
- Optiprep density gradient media (Axis-Shield PoC) (Cosmo Bio, catalog number: AXS-1114542 )
- α-HA antibody (monoclonal anti-HA.11) (BioLegend, catalog number: 901513 )
- Dithiothreitol (DTT)
- Golgi membranes, vesicles, cytosol (see preparation of working aliquots under Recipes)
- Sucrose (Fisher Scientific, catalog number: S/8600/60 )
- HEPES
- Creatine phosphokinase
- GTP
- ATP
- Creatine phosphate
- Potassium hydroxide (KOH)
- Magnesium acetate (Mg(OAc)2)
- Magnesium chloride (MgCl2)
- Trypsin, 2.5% (10x) (Thermo Fisher Scientific, GibcoTM, catalog number: 15090046 )
- Phosphate buffered saline (PBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 10010023 )
- Buffers (see Recipes)
- Assay sucrose
- ATP/GTP mixture (10x)
- Cytosol buffer
- HM buffer
- KHM buffer
- Reaction buffer (10x)
- Trypsin-PBS buffer
- Assay sucrose
Note: All reagents and buffers should be stored in convenient sized aliquots at -80 °C. When running low on critical aliquots (membranes, cytosol, ATP/GTP mixture), prepare a new set and test it against the old ones to ensure reproducibility.
Equipment
- Microwave oven
- Water bath
- Sonicating water bath (Grant Instruments, model number: XUBA3 )
- Incubator
- 1 ml Dounce homogenizer (DWK Life Sciences, Wheaton, catalog number: 357538 )
- Centrifuge
- Ultracentrifuge
- SW41 rotor (Beckman Coulter, model: SW 41 Ti ) and 13.2 ml thinwall ultra-clearTM tubes (Beckman Coulter, catalog number: 344059 )
- Sugar refractometer (range 0-50%) (Bellingham and Stanley Ltd, UK)
- TLA 100.3 rotor (Beckman Coulter, model: TLA-100.3 ) and 3.5 ml thickwall polycarbonate tubes (Beckman Coulter, catalog number: 349622 )
- TLS-55 rotor (Beckman Coulter, model: TLS-55 ) and 2.2 ml thinwall ultra-clearTM tubes (Beckman Coulter, catalog number: 347356 )
- Standard mammalian cell culture apparatus
- Evolve 512 EMCCD (electron multiplying charged coupled device) Camera (Photometrics, model: Evolve® 512 )
- Zeiss Axiovert 200M fully motorized inverted microscope (Carl Zeiss, model: Axiovert 200M )
- X-Cite 120Q excitation light source (Excelitas Technologies, model: X-Cite 120Q )
- CFP filter (Chroma Technology, catalog number: 49001 )
- YFP filter (Chroma Technology, catalog number: 49003 )
- Objective lens (Zeiss Plan-Apochromat 63x/1.40 Oil DIC, Carl Zeiss Ltd, Cambridge, UK)
- Black card to exclude room light from samples during imaging
Software
- ZEN 2009 software (www.zeiss.com)
- PM Capture Pro software (http://www.photometrics.com)
- AutoHotkey (www.autohotkey.com) (AutoHotkey Foundation LLC)
- ImageJ (https://imagej.nih.gov/ij/)
Procedure
- Experimental preparation
- Water
Note: Molecular biology grade water was used for all procedures, including the washing of microscope slides and coverslips. - Microscope slides and coverslips
- To fully submerge the slides and coverslips in their racks, sufficient water was warmed to ~40 °C in a microwave, then the detergent decon90 (Decon Laboratories Limited, Sussex, UK) was added to 3% (v/v). In their racks, slides and coverslips were dipped up and down 10 times in the detergent solution to help dislodge surface contaminants, then left to soak for ~7 h.
- After dipping up and down a further 5 times, the solution was discarded and the glassware rinsed once with water as follows: sufficient water was added to the container to fully submerge glassware which was then dipped up and down 10 times. Fresh 3% (v/v) decon90 solution was made up, and the glassware left to soak in this for at least 12 h. Glassware was then thoroughly rinsed at least 5 times in water, then placed in an oven at 60 °C until dry. Once dry, slides and coverslips were stored in covered boxes to prevent dust contamination.
- To fully submerge the slides and coverslips in their racks, sufficient water was warmed to ~40 °C in a microwave, then the detergent decon90 (Decon Laboratories Limited, Sussex, UK) was added to 3% (v/v). In their racks, slides and coverslips were dipped up and down 10 times in the detergent solution to help dislodge surface contaminants, then left to soak for ~7 h.
- Silica beads
- A suspension containing 5 micron silica beads was prepared for addition to assay mixtures prior to mounting. The beads acted as a spacer between the slide and the coverslip. A small amount of bead powder (~5 μl dry volume) was deposited in a 1.5 ml microfuge tube to which 500 μl of 150 mM KCl, 10 mM HEPES, pH 7.2 was added.
- The beads were distributed in this buffer by vigorous vortexing followed by 30 sec agitation in a sonicating water bath (power 35 W, intensity 44 kHz)–this was repeated once more to make the beads become mono-dispersed. A bead density was required such that 1 μl of this suspension added to a 25 μl assay mixture would provide ~1-2 beads per field of view when 3 μl of the mixture was mounted and viewed under the 63x objective. Therefore, when a new stock of beads was made, the bead density was tested by mixing 1 μl of the stock with 25 μl water and mounting 3 μl of this suspension as described below and viewing under a microscope. If the number of beads was too low, the bead stock was briefly spun down and enough buffer removed to increase bead density when resuspended. More buffer could be added to the bead stock for the opposite effect.
- A suspension containing 5 micron silica beads was prepared for addition to assay mixtures prior to mounting. The beads acted as a spacer between the slide and the coverslip. A small amount of bead powder (~5 μl dry volume) was deposited in a 1.5 ml microfuge tube to which 500 μl of 150 mM KCl, 10 mM HEPES, pH 7.2 was added.
- Mammalian cell culture
Wild type and HEK293 cell clones stably expressing GalT-XFP constructs (Cottam et al., 2014) were used. These were generated by stable transfection of a pCR3.1 vector (Invitrogen) based plasmid containing human β-1,4-galactosyltransferase 1 with a CFP or a YFP tag, and selection with 800 µg/ml Geneticin (GibcoTM). Single cell-derived clones were selected for use if the expressed GalT-XFP was found localized in the Golgi only, rather than partly distributed in cytosolic puncta. Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM GlutaMAX-I, and 100 U of penicillin and 100 μg of streptomycin per ml. Most other cell lines were cultured in the same media, although CHO lines needed Ham’s F12 media supplemented with 5% FBS and the same antibiotics composition as for HEK cell culture. All cell lines were grown in a humidified incubator at 37 °C and 5% CO2. - Golgi membranes
- The following method is adapted from Balch et al. (1984). Typically, two T175 flasks (each 175 cm2) of stably expressing GalT-CFP HEK cells (Cottam et al., 2014) were grown to a confluent monolayer (~7-10 x 107 cells total), harvested, and washed twice in 0.25 M sucrose, 10 mM HEPES, pH 7.4. The cell pellet (~500-600 μl) was resuspended in four times the pellet volume using the same 0.25 M sucrose buffer. The suspension was treated with 25 strokes in a 1 ml tight fitting Dounce homogenizer. The homogenate was flash frozen in liquid nitrogen and then stored at -80 °C for later subcellular fractionation.
- Golgi membranes were isolated from homogenates by floatation on a discontinuous sucrose gradient by centrifugation. Frozen homogenates were rapidly thawed at 37 °C and then maintained on ice. The sucrose concentration of the homogenate was adjusted to 1.4 M by adding ice-cold 2.3 M sucrose, 10 mM HEPES, pH 7.4 at a volume of 1.28-fold to the homogenate volume, and mixed thoroughly by pipetting with a 10 ml serological pipette.
- The mixture was loaded into an SW41 tube to form the bottom layer and then overlaid with 4.94 ml of 1.2 M sucrose, 10 mM HEPES, pH 7.4. These two layers were then overlaid with enough 0.8 M sucrose, 10 mM HEPES, pH 7.4 to fill the tube to its maximum capacity of 12 ml (between 2.5 ml and 3 ml depending on the volume of the bottom layer). The gradient was then centrifuged for 80 min at 39,000 rpm, 4 °C in an SW41 rotor. In a cold room, the turbid band at the 1.2/0.8 M sucrose interface was then drawn off in a minimum volume (~700-800 μl) by manual pipetting from the top of the gradient downwards. This fraction was mixed well, aliquoted into 20 μl volumes in PCR tubes, and flash frozen in liquid nitrogen. The sucrose concentration of the final sample was measured using a 0-50% sugar refractometer (Bellingham and Stanley Ltd, UK). The sucrose concentration was always very close to and slightly above 1 M.
- In the assay, 1 μl from a stock of Golgi isolated in the above fashion gave ~32 particles per field of view by EMCCD imaging.
- The following method is adapted from Balch et al. (1984). Typically, two T175 flasks (each 175 cm2) of stably expressing GalT-CFP HEK cells (Cottam et al., 2014) were grown to a confluent monolayer (~7-10 x 107 cells total), harvested, and washed twice in 0.25 M sucrose, 10 mM HEPES, pH 7.4. The cell pellet (~500-600 μl) was resuspended in four times the pellet volume using the same 0.25 M sucrose buffer. The suspension was treated with 25 strokes in a 1 ml tight fitting Dounce homogenizer. The homogenate was flash frozen in liquid nitrogen and then stored at -80 °C for later subcellular fractionation.
- Vesicles
- The following method is adapted from Love et al. (1998). Typically, four T175 flasks of stably expressing GalT-YFP HEK cells were grown to a confluent monolayer (~14-20 x 107 cells total), harvested, and washed once in PBS and then in 0.2 M sucrose, 10 mM Tris, pH 7.2. The pellet (~1 ml) was resuspended in four times the pellet volume using the same 0.2 M sucrose buffer, flash frozen in liquid nitrogen and then stored at -80 °C until needed.
- Vesicle membranes were isolated by floatation on a discontinuous gradient of Optiprep density gradient media by centrifugation. Frozen cell suspensions were gradually thawed in a water bath at 21 °C. This caused permeabilization of cells by membrane fracture.
- The cells were centrifuged for 5 min at 1,000 x g, 4 °C, which left vesicles in the supernatant. This supernatant was centrifuged once more for 5 min at 1,000 x g, 4 °C then twice more for 20 min at 20,000 x g, 4 °C to remove all debris and large cellular membranes. Vesicles in the supernatant were then pelleted by ultracentrifugation at 55,000 rpm for 45 min at 4 °C in a TLA 100.3 rotor. The vesicle pellet (typically ~30 μl) was resuspended in KHM buffer (see Recipes) to a volume of 320 μl. This 320 μl was mixed with 480 μl of 50% Optiprep in HM buffer (see Recipes). The mixture was transferred to a TLS-55 tube and overlayed with 800 μl of 25% Optiprep in KHM buffer and then with 400 μl of 10% Optiprep in KHM buffer.
- Vesicles were floated to the 10%/25% interface by ultracentrifugation at 55,000 rpm for 3 h 10 min at 4 °C in a TLS-55 rotor. The turbid band at the interface was harvested by manual pipetting in a minimum volume of typically 300-400 μl and mixed well. 10-15 μl aliquots were made into PCR tubes which were flash frozen in liquid nitrogen. In the assay, 1 μl from a stock of vesicles isolated in the above fashion gave ~150-250 particles per field of view by EMCCD imaging.
- A larger, more concentrated batch of vesicles was produced for studies requiring cytosol dependence by using a scaled-up version of the above method. ~3.5 x 109 GalT-YFP HEK cells were harvested. The Optiprep gradient was increased 6.25 times to 12.5 ml total volume in an SW41 tube, then ultracentrifuged at 41,000 rpm for 8 h at 4 °C. 250 μl fractions were manually drawn off the gradient by pipetting. Western blot analysis of the fractions (using an α-HA antibody against an HA tag in the GalT-YFP construct for detection) showed six consecutive fractions around the 10%/25% interface region which had the highest signal (indicating vesicles). These fractions were pooled (1.5 ml total), aliquoted and flash frozen as above. 1 μl from this concentrated stock of vesicles in the assay gave ~1,000-1,500 particles per field of view by EMCCD imaging.
- The following method is adapted from Love et al. (1998). Typically, four T175 flasks of stably expressing GalT-YFP HEK cells were grown to a confluent monolayer (~14-20 x 107 cells total), harvested, and washed once in PBS and then in 0.2 M sucrose, 10 mM Tris, pH 7.2. The pellet (~1 ml) was resuspended in four times the pellet volume using the same 0.2 M sucrose buffer, flash frozen in liquid nitrogen and then stored at -80 °C until needed.
- Cytosol
- The method for cytosol preparation was adapted from (Balch et al., 1984). Four T175 flasks of cells were grown to full confluency (~14-20 x 107 HEK293 cells in total). Cells were trypsinized using trypsin-PBS buffer, then washed 4 times with PBS before washing twice in ice-cold 0.25 M sucrose, 10 mM HEPES, pH 7.4. The pellet was resuspended in 1.5 ml of the same buffer with fresh DTT added to 1 mM from a 1 M aqueous stock.
- The suspension was treated with 30 strokes in a 1 ml tight fitting Dounce homogenizer. The homogenate was transferred to a TLA 100.3 ultracentrifuge tube and centrifuged at 60,000 rpm for 45 min at 4 °C. The supernatant was removed and centrifuged again in the same way to remove all cell debris. Meanwhile, a PD-10 desalting column was equilibrated with 100 mM KCl, 1 mM DTT, 10 mM HEPES, pH 7.2. The cleared cytosol supernatant was added to the column. The desalted cytosol was eluted with the same equilibration buffer. Most of the protein was contained within the first 2.5 ml of eluate as determined by Bradford protein assay. Protein concentration was typically 7.5-8.5 mg/ml. The eluates were mixed well then aliquoted and flash frozen in liquid nitrogen.
- The method for cytosol preparation was adapted from (Balch et al., 1984). Four T175 flasks of cells were grown to full confluency (~14-20 x 107 HEK293 cells in total). Cells were trypsinized using trypsin-PBS buffer, then washed 4 times with PBS before washing twice in ice-cold 0.25 M sucrose, 10 mM HEPES, pH 7.4. The pellet was resuspended in 1.5 ml of the same buffer with fresh DTT added to 1 mM from a 1 M aqueous stock.
- Water
- Set-up, incubation and mounting
- Set-up
Note: All samples, except for water, must be kept on ice that is mixed with cold water (to allow for good heat exchange) prior to use in the assay.- Set the water bath to 37 °C. Prepare dilutions of all other reagents before defrosting membranes, cytosol and ATP/GTP mixture.
Note: Defrost membrane aliquots and cytosol quickly in your hand and then put on wet ice. - Aliquots of vesicles, Golgi, cytosol and ATP must be briefly spun down (~3 sec) in a microfuge straight after defrosting.
Note: Do not vortex or flick membranes, cytosol or ATP/GTP, rather mix these by setting pipette volume to ~10% less than the aliquot volume and pipet up and down 7 times before any volume is removed. Buffers, in contrast, should be mixed by vortexing after defrosting. - Dilute the vesicle stock to ensure about 70-150 vesicles per image.
Notes: - This should be tested for new vesicle preps, but is generally in the range of a 10-30x dilution.
- Assay mixtures should be made up in 0.5 ml microfuge tubes. Mix assay components in the order: water, reaction buffer and other buffers, cytosol, Golgi, vesicles, and ATP/GTP mixture.
- Set the water bath to 37 °C. Prepare dilutions of all other reagents before defrosting membranes, cytosol and ATP/GTP mixture.
- Incubation
- A typical assay will contain: 3.8 µl Golgi (see Note 1), 4 µl diluted vesicle stock, cytosol equivalent to 57 mg total protein, cytosol buffer (see Recipes) such that the sum of this and the cytosol volume is 15.5 µl, 5 µl 10x concentrated reaction buffer (see Recipes), 2.75 µl assay sucrose (see Recipes), 5 µl 10x concentrated ATP/GTP mixture (see Recipes) in a 50 µl total volume. The assay mixture is gently pipetted up and down 10 times before placing 25 µl into a separate tube. Half of the assay mixture is left on wet ice while the other is transferred to the 37 °C water bath.
- Samples are incubated at 37 °C for 40 min in the dark, then transferred back onto wet ice. Assay mixtures are mounted immediately after incubation.
- A typical assay will contain: 3.8 µl Golgi (see Note 1), 4 µl diluted vesicle stock, cytosol equivalent to 57 mg total protein, cytosol buffer (see Recipes) such that the sum of this and the cytosol volume is 15.5 µl, 5 µl 10x concentrated reaction buffer (see Recipes), 2.75 µl assay sucrose (see Recipes), 5 µl 10x concentrated ATP/GTP mixture (see Recipes) in a 50 µl total volume. The assay mixture is gently pipetted up and down 10 times before placing 25 µl into a separate tube. Half of the assay mixture is left on wet ice while the other is transferred to the 37 °C water bath.
- Mounting
Prior to mounting, the silica bead suspension was briefly vortexed, and then 1 µl of this added to the assay mixture. The assay mixture was pipetted up and down 20 times with a pipet set to 21 µl, then quickly 3 µl was applied as two separate drops onto one half of a slide and immediately covered with a clean square coverslip. The edges were quickly sealed with clear nail varnish to prevent sample evaporation and fix in place. The 37 °C and ice incubated halves of the same assay were mounted in pairs onto the same slide. Slides were stored protected from light in a 4 °C fridge until imaging. Figure 1 shows how sample mounting is performed in the overall context of how the assay is executed.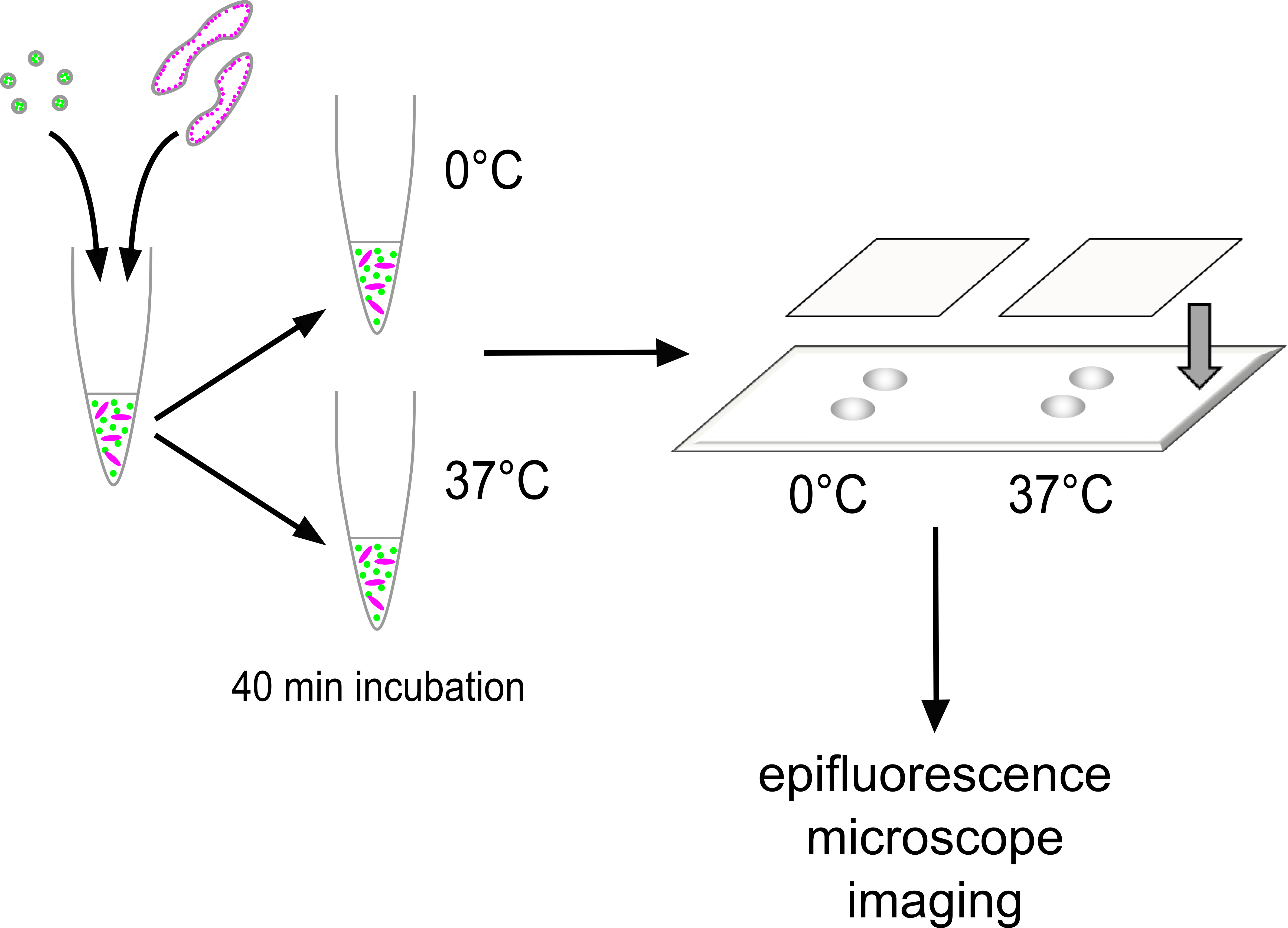
Figure 1. Execution of the assay. GalT-YFP vesicles and GalT-CFP Golgi are mixed together in a total reaction volume of 50 μl containing the desired reaction conditions. The mixture is equally divided into two tubes. One is incubated at 0 °C for 40 min as an internal control, and the other is incubated at 37 °C for the same period. After incubation, 5 micron silica beads are mixed with each sample, then 3 μl of the mixture is immediately delivered in two roughly equally-sized spots onto a microscope slide. The sample is covered with a 22 x 22 mm coverslip, sealed around the edges with clear nail varnish with the silica beads acting as spacers. The slide is kept in the dark at 4 °C until imaging by epifluorescence microscopy at room temperature.
- Set-up
- Imaging
- Equipment set-up and control
- Images were collected by bright-field microscopy using an Evolve 512 EMCCD camera, attached to a ZEISS Axiovert 200M fully motorized inverted microscope. Illumination was provided by an X-Cite 120Q excitation light source. The adjustable iris of the X-Cite was set to level 3 of 4, where level 4 is fully open for maximum output. The objective lens was a ZEISS Plan-Apochromat 63x/1.40 Oil DIC. The microscope was controlled by ZEN 2009 software (ZEISS) and the camera was controlled by PM Capture Pro software (Photometrics) running on the same computer system as the ZEISS software. CFP and YFP filters from Chroma Technology Corporation were used. Although confocality is not used, it is essential to use a fully motorized stage, filter wheel and shutters, hence the use of this microscope. Extraneous room light on the sample during imaging was minimized by using low-light conditions and by also shielding the sample with matt black card as follows. On a circular disc of the card (~7 cm in diameter), a circular hole was cut in the center that was slightly smaller than the diameter of the outer casing of the retractable front lens on the 63x objective. The front lens was gently pushed through the hole so that the disc formed a wide ‘collar’ to block light from below, then a sheet of card with a central rectangular hole was placed over the objective to cover gaps. Subsequently the universal mounting frame (i.e., the part which clips into the motorized stage) was placed above the rectangular card to hold the slide. On the microscope stage, a box lid made of the same card of dimensions approximately 15 x 10 x 2 cm (L x W x H), was placed over the sample (Figure 2).

Figure 2. Shielding the mounted sample during imaging. Although low-light room conditions are used during imaging, extraneous light on the sample is also minimized by shielding. Matt black card is utilized as follows: 1. a wide collar is fitted around the objective lens to block light from below; 2. a sheet with a rectangular hole is placed over the objective to cover gaps; and 3. a shallow box lid is placed on top to block light from above. The slide is placed in the universal mounting frame on top of the rectangular sheet in image 2. - The open source software AutoHotkey was used to write scripts to automate control of the ZEN 2009 and PM Capture Pro software. The scripts (programmed to be launched by pressing the keyboard F-keys or buttons of a joypad i.e., as ‘hotkeys’) moved the cursor to activate commands in both the microscope and camera software windows with appropriate time delays. The scripts were programmed so that the operations of changing filters, opening/closing the lamp shutter, and camera image acquisition occurred in sequence to obtain 1 image each for vesicles (YFP channel) and Golgi (CFP channel). This ensured all images were captured under the same conditions, and without unnecessary delay to avoid bleaching of the fluorophores.
- Images were collected by bright-field microscopy using an Evolve 512 EMCCD camera, attached to a ZEISS Axiovert 200M fully motorized inverted microscope. Illumination was provided by an X-Cite 120Q excitation light source. The adjustable iris of the X-Cite was set to level 3 of 4, where level 4 is fully open for maximum output. The objective lens was a ZEISS Plan-Apochromat 63x/1.40 Oil DIC. The microscope was controlled by ZEN 2009 software (ZEISS) and the camera was controlled by PM Capture Pro software (Photometrics) running on the same computer system as the ZEISS software. CFP and YFP filters from Chroma Technology Corporation were used. Although confocality is not used, it is essential to use a fully motorized stage, filter wheel and shutters, hence the use of this microscope. Extraneous room light on the sample during imaging was minimized by using low-light conditions and by also shielding the sample with matt black card as follows. On a circular disc of the card (~7 cm in diameter), a circular hole was cut in the center that was slightly smaller than the diameter of the outer casing of the retractable front lens on the 63x objective. The front lens was gently pushed through the hole so that the disc formed a wide ‘collar’ to block light from below, then a sheet of card with a central rectangular hole was placed over the objective to cover gaps. Subsequently the universal mounting frame (i.e., the part which clips into the motorized stage) was placed above the rectangular card to hold the slide. On the microscope stage, a box lid made of the same card of dimensions approximately 15 x 10 x 2 cm (L x W x H), was placed over the sample (Figure 2).
- Sample handling and image acquisition
- The sample should be removed from the fridge a couple minutes before imaging to stop condensation build-up. Once on the microscope, the room must be switched to low-light conditions and the sample must be shielded from external light by covering it with matt black card from top and bottom as described above. Focusing is tricky due to the lack of contrast of the membranes under bright-field illumination. Initial focusing should aim for the 5 µm glass beads using the halogen lamp on low intensity.
- Then, for finer focus using the YFP fluorescence of the vesicles, the high gain preview mode of the EMCCD camera must be used (see Note 2). They are too faint to be seen via the eyepiece and the camera needs to ‘see’ the precise focal plane of these sub-micron particles. It is easy to overfocus, thereby squashing the sample, in which case it has to be re-mounted. Once the focal plane with the vesicles is found, care should be taken so that all images are collected from the particles sitting on the coverslip rather than the ones on the slide. A quick refocusing away from the coverslip will verify this by showing the slide-attached particles; this check should be performed regularly. Moving the distance of a few fields of view in the x-y plane usually keeps the focus close enough to require only little adjustments to regain focus.
- For each assay condition, 12-16 images per incubation temperature need to be collected. Particles bleach quickly, so care should be taken that the focusing time overall does not exceed 7 sec before image acquisition is started. Camera settings may need to be adjusted depending on the age of the excitation lamp. A set of ideal settings are in Note 2. A very short exposure time with binning and a high gain is used for finding an appropriate area and focusing. Conversely, a long exposure time without binning and low gain is used to acquire images of vesicles and Golgi with higher resolution and lower noise. Scripts need to be written (using for example the AutoHotkey program for PC) to automate the sequence of operations performed by the microscope and camera to acquire images for the YFP and CFP channels (described above). These scripts should be programmed to launch upon pressing single ‘hotkeys’ such as the F-keys, or even the buttons of a joypad can be assigned. This allows single click initiation of the three presets (see Note 2) that allow preview mode for focusing, followed by YFP and CFP image acquisition.
Note: We found that programming further keys for saving the YFP (vesicles) and CFP (Golgi) channel images with a V_ or G_ prefix followed by a time-stamp in the file name made data acquisition and subsequent processing much more user-friendly. This also ensured that the correct vesicle and Golgi images from the same field of view were always paired up together afterwards.
- The sample should be removed from the fridge a couple minutes before imaging to stop condensation build-up. Once on the microscope, the room must be switched to low-light conditions and the sample must be shielded from external light by covering it with matt black card from top and bottom as described above. Focusing is tricky due to the lack of contrast of the membranes under bright-field illumination. Initial focusing should aim for the 5 µm glass beads using the halogen lamp on low intensity.
- Equipment set-up and control
Data analysis
- ImageJ was used to process and analyze all images. For Golgi, images were converted to 8 bit, then background noise reduced using the ‘subtract background’ command (rolling ball radius 2.0 pixels, sliding paraboloid). The threshold was set to ‘Triangle dark’, and the lower threshold value was multiplied by a factor of 1.1 to ensure that particles were selected above background noise. A binary image was made of the selection, and any selected particles containing holes were filled in. A summary of the number of particles was generated. The Look Up Table for the binary image was changed to ‘red’ and saved. The same process was conducted for vesicle images except that after the ‘Triangle dark’ threshold, the lower threshold value was multiplied by a factor of 1.4. The Look Up Table for the binary image was changed to ‘green’ and saved. The multiplying factor, for both Golgi and vesicles images, can be increased in case images are very noisy, or even decreased if noise is low. However, the same factor should be used for all images collected on the same day. Both the Golgi (red) and vesicle (green) binary images were opened, converted to RGB color images, and then merged into a composite image where overlapping red and green particles were additive to give yellow pixels. Using the ‘connection thresholding’ plugin (obtained from http://imagejdocu.tudor.lu), the thresholds in the dialogue box for this plugin were adjusted so that Golgi were selected in blue, overlapping (colocalized) pixels in red, and non-colocalizing vesicle particles were excluded. The ‘Hyst’ (Hysteresis) command in the dialogue box was selected. This caused any Golgi particle containing one or more colocalized pixels to be filled in as a single colocalization event, and output as a binary image. A threshold of ‘255, 255’ was applied and the number of such colocalization events was counted. These image processing operations are summarized in Figure 3. The above operations carried out on Golgi and vesicle images were compiled into ImageJ macros (Note 3) which allowed multiple images to be processed as a batch. Following the processing, all images should be looked at to remove false positive colocalization events caused by the 5 µm beads, which occasionally give fluorescent signals in both channels, but can easily be distinguished from real events due to the shape of the putative particle.
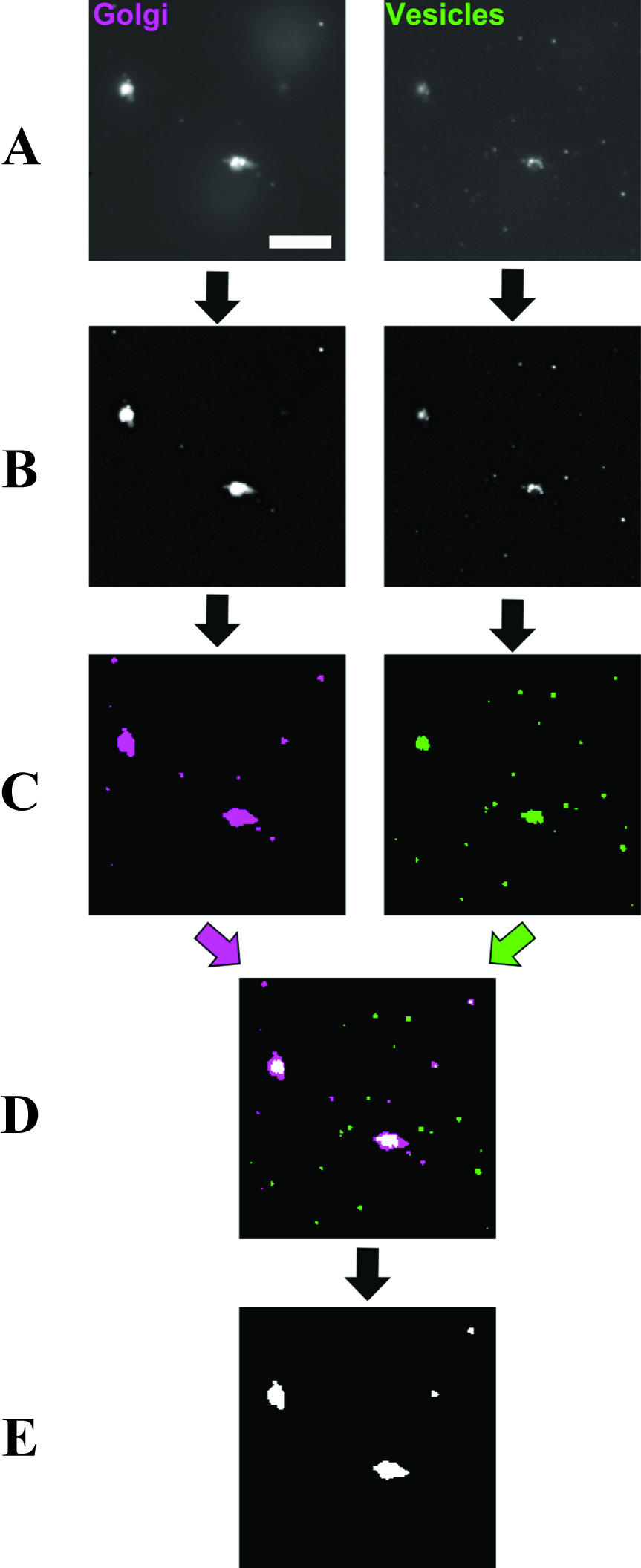
Figure 3. Image processing workflow (Cottam, 2012). A. Raw images of Golgi and vesicles are treated with background subtraction. This also corrects slight uneven illumination of the sample generating the images in (B). B. An intensity threshold is applied to images to select particles above the average background noise. C. The selection is converted to a binary image and re-colored to magenta and green for Golgi and vesicles respectively. D. The binary images are overlaid to reveal colocalized areas as white pixels. E. A hysteresis operation is applied to the overlaid image which completely fills any particle containing white pixels to become a single colocalization event. The number of colocalization events is expressed as a percentage of the total number of particles (colocalization events/(Golgi + vesicles)) to give the measure of assay activity. In our previous work, we have used the calculation (Golgi + vesicles + colocalization events) for total particle numbers, which is not correct but should be considered when comparing data to our published results. Given the generally low number of colocalization events (Figure 4), the distortion by the incorrect particle total does not change the biological conclusions when comparing results for different assay conditions. Scale bar = 10 μm. - In Excel, the number of colocalized particles was expressed as a percentage of the total number of particles ((colocalized/(Golgi + vesicles)); see explanation in the legend of Figure 3). This gave the percentage of activity in the sample. Activity for the 37 °C incubation within each assay condition was then normalized to the activity in the corresponding ice control, giving the normalized activity of each condition (i.e., the fold increase compared to the ice control) (see Figure 4). Background activity does vary from day to day. However, we found that it was not practical to perform assays with more than six separate conditions on one day. Therefore a further normalization step was introduced, in which each condition’s normalized activity was further normalized to a wild type (or other suitable) control sample measured on the day. Activities normalized in this manner to the same control on different measurement days could be robustly compared and averaged.
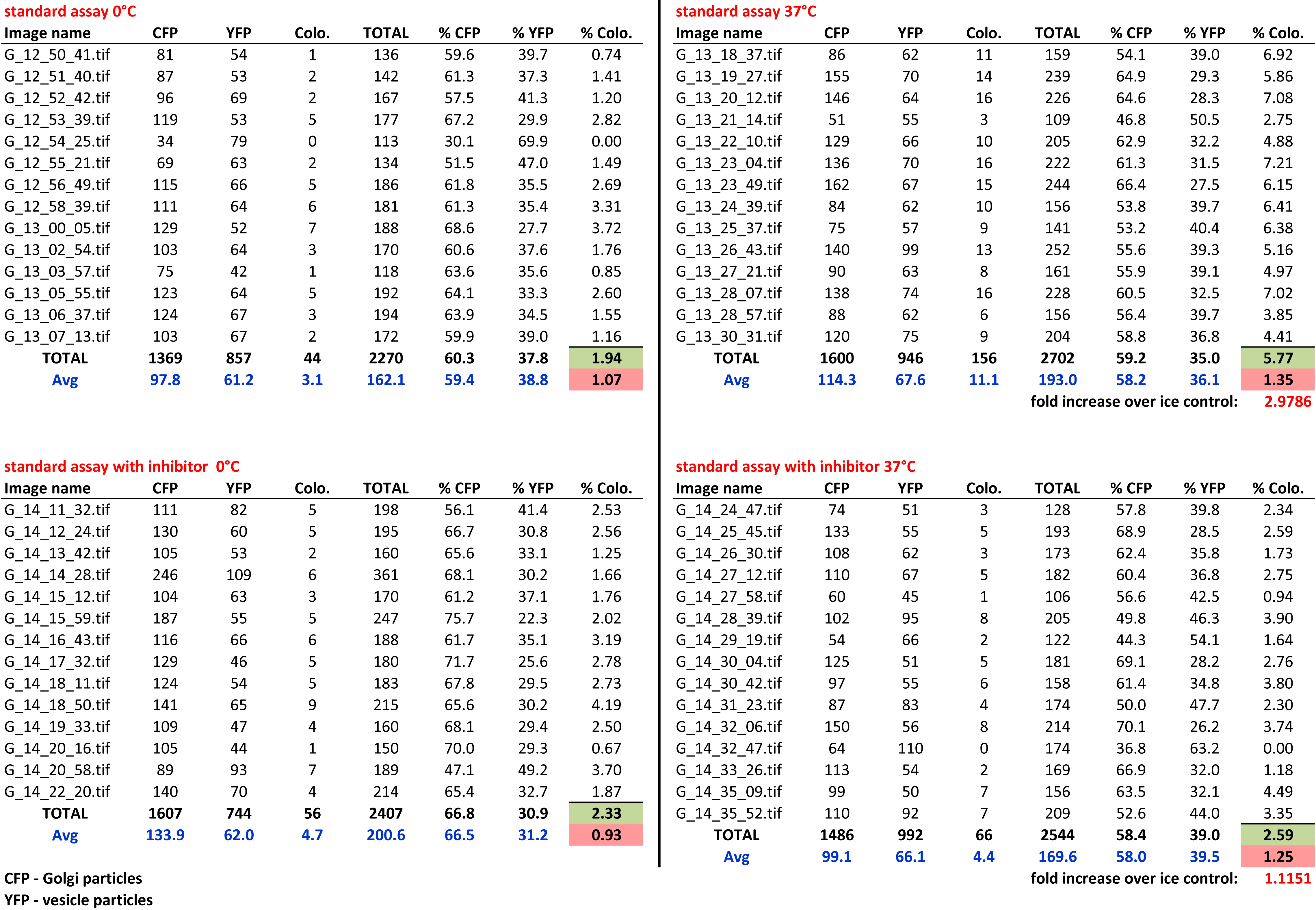
Figure 4. Typical data sheet of the image processing results. Particle numbers as well as the numbers of co-localizing particles are shown for each image.
Notes
- When following the preparation protocol above, the concentration of the prep allows the amount of Golgi membranes in the assay to be very close to 3.8 µl giving about 70-150 particles per image. If the concentration of the Golgi preparation is very different, care needs to be taken to balance the amount of Golgi added with the sucrose buffer such that the osmolarity of the final assay mixture is not too far off the 310 mOsm of DMEM.
- Camera preset parameters in the PM Capture Pro software:
- Preset 1–preview mode (high gain, high noise) for finding the right area and focusing quickly:
2 x 2 binning, analogue-to-digital gain 3, exposure 120 msec, electron multiplication gain = 220 - Preset 2–YFP capture (low gain, low noise):
1 x 1 binning, analogue-to-digital gain 3, exposure 4.5 sec, electron multiplication gain = 22 - Preset 3–CFP capture (low gain, low noise):
1 x 1 binning, analogue-to-digital gain 3, exposure 2.6 sec, electron multiplication gain = 8
- Preset 1–preview mode (high gain, high noise) for finding the right area and focusing quickly:
- Macros for ImageJ
- YFP vesicle processing:
for (i=1; i<=nImages; i++) {
selectImage(i);
run("8-bit");
run("Subtract Background...", "rolling=2 sliding");
setAutoThreshold("Triangle dark");
getThreshold(lower, upper);
setThreshold(lower*1.4, 255);
run("Make Binary", "thresholded remaining black");
run("Invert");
run("Fill Holes");
setThreshold(255, 255);
run("Analyze Particles...", "size=0-infinity circularity=0.00-1.00 show=Nothing clear include summarize");
run("Green");
}
run("Images to Stack", "name=YFP_stack stack title=[] use");
saveAs("Tiff") - CFP Golgi processing:
for (i=1; i<=nImages; i++) {
selectImage(i);
run("8-bit");
run("Subtract Background...", "rolling=2 sliding");
setAutoThreshold("Triangle dark");
getThreshold(lower, upper);
setThreshold(lower*1.1, 255);
run("Make Binary", "thresholded remaining black");
run("Invert");
run("Fill Holes");
setThreshold(255, 255);
run("Analyze Particles...", "size=0-infinity circularity=0.00-1.00 show=Nothing clear include summarize");
run("Red");
}
run("Images to Stack", "name=CFP_stack stack title=[] use");
saveAs("Tiff") - To generate a merged RGB stack:
for (i=1; i<=nImages; i++) {
selectImage(i);
run("RGB Color");
}
run("Merge Channels...", "red=CFP_stack.tif green=YFP_stack.tif blue=*None* gray=*None* create");
saveAs("Tiff")
- YFP vesicle processing:
Recipes
- Assay sucrose
1.2 M sucrose
10 mM HEPES, pH 7.4 - ATP/GTP mixture (10x)
Creatine phosphokinase 1,500 U/ml
10 mM GTP
5 mM ATP
200 mM creatine phosphate
7.5 mM KOH to neutralise the ATP
20 mM HEPES, pH 7.4 - Cytosol buffer
100 mM KCl
1 mM DTT
10 mM HEPES, pH 7.2 - HM buffer
10 mM HEPES, pH 7.2
2.5 mM Mg(OAc)2 - KHM buffer
150 mM KCl
2.5 mM Mg(OAc)2
10 mM HEPES, pH 7.2 - Reaction buffer (10x)
250 mM HEPES, pH 7.4
20 mM MgCl2 - Trypsin-PBS buffer
0.25% (v/v) trypsin in PBS
Acknowledgments
We are grateful to the Imaging Facility at the University of York, Department of Biology’s Bioscience Technology Facility. This work was supported by a BBSRC PhD studentship supporting NPC awarded to DU and Marie Curie grant (201098) to DU. The protocol was published in (Cottam et al., 2014). The authors declare that there’s no conflicts of interest.
References
- Balch, W. E., Dunphy, W. G., Braell, W. A. and Rothman, J. E. (1984). Reconstitution of the transport of protein between successive compartments of the Golgi measured by the coupled incorporation of N-acetylglucosamine. Cell 39(2 Pt 1): 405-416.
- Cottam, N. P. (2012). A cell-free vesicle tethering assay. PhD thesis, University of York.
- Cottam, N. P. and Ungar, D. (2012). Retrograde vesicle transport in the Golgi. Protoplasma 249(4): 943-955.
- Cottam, N. P., Wilson, K. M., Ng, B. G., Korner, C., Freeze, H. H. and Ungar, D. (2014). Dissecting functions of the conserved oligomeric Golgi tethering complex using a cell-free assay. Traffic 15(1): 12-21.
- Fisher, P., and Ungar, D. (2016). Bridging the gap between glycosylation and vesicle traffic. Front Cell Dev Biol 4: 15.
- Kingsley, D. M., Kozarsky, K. F., Segal, M. and Krieger, M. (1986). Three types of low density lipoprotein receptor-deficient mutant have pleiotropic defects in the synthesis of N-linked, O-linked, and lipid-linked carbohydrate chains. J Cell Biol 102(5): 1576-1585.
- Love, H. D., Lin, C. C., Short, C. S. and Ostermann, J. (1998). Isolation of functional Golgi-derived vesicles with a possible role in retrograde transport. J Cell Biol 140(3): 541-551.
- Luebbehusen, J., Thiel, C., Rind, N., Ungar, D., Prinsen, B. H., de Koning, T. J., van Hasselt, P. M. and Koerner, C. (2010). Fatal outcome due to deficiency of subunit 6 of the conserved oligomeric Golgi complex leading to a new type of congenital disorders of glycosylation. Hum Mol Genet 19(18): 3623-3633.
- Soldati, T., Riederer, M. A. and Pfeffer, S. R. (1993). Rab GDI: a solubilizing and recycling factor for rab9 protein. Mol Biol Cell 4(4): 425-434.
- Ungar, D., and Hughson, F. M. (2003). SNARE protein structure and function. Annu Rev Cell Dev Biol 19: 493-517.
- Wu, X., Steet, R. A., Bohorov, O., Bakker, J., Newell, J., Krieger, M., Spaapen, L., Kornfeld, S. and Freeze, H. H. (2004). Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat Med 10(5): 518-523.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Cottam, N. P. and Ungar, D. (2017). Cell-free Fluorescent Intra-Golgi Retrograde Vesicle Trafficking Assay. Bio-protocol 7(22): e2616. DOI: 10.21769/BioProtoc.2616.
Category
Biochemistry > Protein > Activity
Cell Biology > Organelle isolation > Golgi
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.