- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Uptake Assays in Xenopus laevis Oocytes Using Liquid Chromatography-mass Spectrometry to Detect Transport Activity
Published: Vol 7, Iss 20, Oct 20, 2017 DOI: 10.21769/BioProtoc.2581 Views: 10562
Reviewed by: Tie LiuBin TianAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Xenopus laevis oocytes are a widely used model system for characterization of heterologously expressed secondary active transporters. Historically, researchers have relied on detecting transport activity by measuring accumulation of radiolabeled substrates by scintillation counting or of fluorescently labelled substrates by spectrofluorometric quantification. These techniques are, however, limited to substrates that are available as radiolabeled isotopes or to when the substrate is fluorescent. This prompted us to develop a transport assay where we could in principle detect transport activity for any organic metabolite regardless of its availability as radiolabeled isotope or fluorescence properties.
In this protocol we describe the use of X. laevis oocytes as a heterologous host for expression of secondary active transporters and how to perform uptake assays followed by detection and quantification of transported metabolites by liquid chromatography-mass spectrometry (LC-MS). We have successfully used this method for identification and characterization of transporters of the plant defense metabolites called glucosinolates and cyanogenic glucosides (Jørgensen et al., 2017), however the method is usable for the characterization of any transporter whose substrate can be detected by LC-MS.
Background
Oocytes from the African clawed frog (Xenopus laevis) is a well-established expression system for heterologous expression and characterization of membrane proteins (i.e., transporters and channels). The X. laevis oocyte express few endogenous membrane proteins and has a low background transport activity. Furthermore, secondary active transporters from plants (Boorer et al., 1992; Theodoulou and Miller, 1995; Nour-Eldin et al., 2006), animals (Sumikawa et al., 1981; Sigel, 1990) and microbes (Calamita et al., 1995; Wahl et al., 2010) have been successfully expressed in X. laevis oocytes, showing that this system is widely applicable to characterize transporters from any organism.
A transport assay requires the expression of the transport protein in a system capable of folding the protein correctly and localizing it to a membrane across which movement of substrate can be detected. Due to the minute amounts moved, researchers have typically used radiolabeled substrates for transport assays. By washing oocytes after incubation and scintillation counting of the oocytes interior accumulation of substrate inside the oocyte could be detected. We have previously utilized this method to identify and characterize sucrose and glucose transporters from Arabidopsis thaliana using the Xenopus oocytes system (Nour-Eldin et al., 2006; Norholm et al., 2006). However, for identification and characterization of plant specialized metabolite transporters, it can be very challenging to generate radiolabeled isotopes of a target substrate. To overcome this challenge we developed a protocol for detecting and quantifying transport of specialized metabolites into X. laevis oocytes by use of LC-MS. Use of this method has allowed us to expand the inventory of assayable substrates to anything that can be detected and quantified by the LC-MS system applied.
Materials and Reagents
- Pipette filter tips (e.g., Biotix, catalog numbers: M-0012-9FC , M0020-9FC , M-0300-9FC , M-1250-9FC96 )
- Petri dishes for oocyte washing (e.g., 90 mm diameter, Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 263991 )
- 24-well NuncTM cell-culture dish (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 142475 )
- Pasteur pipette for oocyte handling
- 1.5 ml tubes (e.g., 1.5 ml microfuge tubes, ‘Easy Fit’, Almeco, catalog number: 02.023.01001 )
- 0.22 μm PVDF-based filter plate (EMD Millipore, catalog number: MSGVN2250 )
- LC-MS vials
- Oocytes expressing transporter(s) of interest and water-injected control oocytes (see Jørgensen et al., 2016 for protocols on making and injecting cRNA and handling oocytes)
Notes:- A high-affinity glucosinolate transporter from Arabidopsis thaliana, ARABIDOPSIS THALIANA GLUCOSINOLATE TRANSPORTER-1 (AtGTR1) (UniProt, catalog number: Q944G5), is used as an example here.
- Oocytes can be purchased from Ecocyte-biosciences (http://ecocyte-us.com/).
- A high-affinity glucosinolate transporter from Arabidopsis thaliana, ARABIDOPSIS THALIANA GLUCOSINOLATE TRANSPORTER-1 (AtGTR1) (UniProt, catalog number: Q944G5), is used as an example here.
- Substrate
Note: We use 4-methylthiobutyl glucosinolate (4MTB) and 3-indolylmethylglucosinolate (I3M) obtained from C2 Bioengineering (http://www.glucosinolates.com/) and CFM Oskar Tropitzsch GmbH, Marktredwitz (http://www.cfmot.de/), respectively. Cyanogenic glucoside linamarin can be purchased from Santa Cruz Biotechnology.
- Methanol for HPLC ≥99.9% (Sigma-Aldrich, catalog number: 34860 )
- Formic acid, reagent grade (Sigma-Aldrich, catalog number: F0507 )
- Acetonitrile for HPLC (Sigma-Aldrich, catalog number: 34851 )
- Sodium chloride (NaCl) (Duchefa Biochemie, catalog number: S0520.5000 )
- Potassium chloride (KCl) (Merck, catalog number: 1.04936.1000 )
- Magnesium chloride hexahydrate (MgCl2·6H2O) (Sigma-Aldrich, catalog number: M2670 )
- Calcium chloride dihydrate (CaCl2·2H2O) (EMD Millipore, catalog number: 1.02382 )
- HEPES (Sigma-Aldrich, catalog number: H4034 )
- Tris-HCl solutions (Trisma hydrochloride) (Sigma-Aldrich, catalog number: T3253 )
- MES (Sigma-Aldrich, catalog number: M8250 )
- Kulori media (pH 7.4) (see Recipes)
- Kulori media (pH 5.6) (see Recipes)
Equipment
- 17 °C incubator for oocyte storage (BINDER, model: Model KB 23 )
- Pipettes (10 µl, 50 µl, 200 µl and 1,000 µl)
- Table top centrifuge for 1.5 ml microfuge tubes (e.g., Ole Dich Instrumentmakers, model: Ole Dich 157 , max speed 20,000 x g, refrigerated)
- Kinetex 1.7u XB-C18 column (100 x 2.1 mm, 1.7 μm, 100 Å) (Phenomenex, catalog number: 00D-4499-AN )
- LC-MS Triple Quadrupole system for sample analysis/data acquisition (Advance UHPLC coupled to an EVOQ Elite Triple Quadrupole mass spectrometer) (Bruker, model: EVOQ EliteTM Triple Quadrupole )
Software
- Microsoft Excel for data analysis and presentation
- Bruker MS Workstation software (Version 8.2, Bruker, Bremen, Germany)
Procedure
In the following, we will provide an example of a typical assay where we test the uptake activity of a given transport protein towards glucosinolates. We test seven oocytes that have been injected with cRNA for our transporter of interest and seven oocytes which have been injected with water to be used as negative control. A set of negative control oocytes should be included for every compound to be tested.
- Assay preparation
- Prepare seven oocytes expressing the transporter of interest (injected with Complimentary RNA (cRNA) [25-50 ng] and incubated at 17 °C for 72 h prior) and seven oocytes that have been injected with water to be used as control oocytes (and incubated at 17 °C for 72 h prior). Injection volume is typically 50 nl.
Note: See Jørgensen et al., 2016 for a detailed protocol for cRNA generation and injection and for oocyte handling from injection to assay.
- 24-well NuncTM cell-culture dish plates are prepared as shown in Figure 1. The pre-incubation well (Figure 1B) is filled with 2 ml Kulori media (pH 5.6, see Recipes). The assay well (Figure 1C) is filled with 1 ml Kulori media (pH 5.6) with substrate (for AtGTRs typically between 100 µM and 1 mM glucosinolate to measure uptake in the high-affinity range).
Note: A mastermix of Kulori pH 5.6 media and substrate is prepared so exactly that the same concentration of substrate is found in each well.
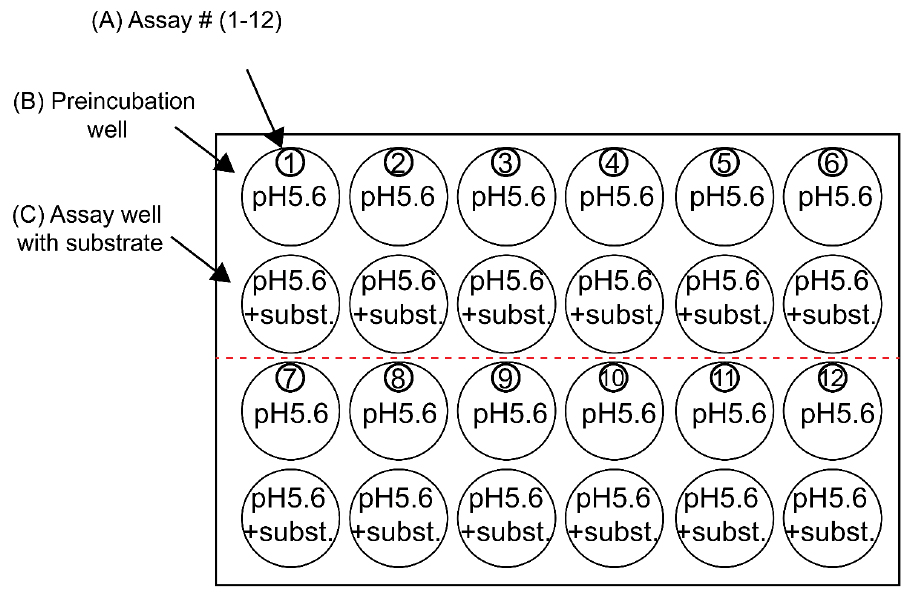
Figure 1. 24-well assay plates suitable for 12 assays at a time
- Three Petri dishes are filled with cold (4 °C) Kulori media (pH 7.4, see Recipes) and stored in fridge.
- Number two sets of 12 microfuge tubes from 1-12. Tubes numbered 1-6 will be used for samples from oocytes expressing the transporter. Tubes numbered 7-12 will be used for the samples from the control oocytes.
Note: Use different colours for the two sets of tubes (e.g., the first set of 12 tubes are labelled with a black marker and the second set is marked with a red marker).
- To keep track of the assay during the experiment and for laboratory book reporting, we recommend using an assay schematic as shown in Table 1.
Table 1. Assay schematic

- Prepare seven oocytes expressing the transporter of interest (injected with Complimentary RNA (cRNA) [25-50 ng] and incubated at 17 °C for 72 h prior) and seven oocytes that have been injected with water to be used as control oocytes (and incubated at 17 °C for 72 h prior). Injection volume is typically 50 nl.
- Assay
- To start the first transport assay, preincubate 6-7 oocytes expressing AtGTR1 in the preincubation well containing Kulori media (pH 5.6) for 5 min (Figure 1B). Subsequently, use a Pasteur pipette to transfer oocytes to the assay well containing Kulori media (pH 5.6) with substrate (Figure 1C) and incubate for 2-180 min at room temperature. Make sure to transfer oocytes in only one drop from the pipette (see Figure 2). Wait three minutes and start the next transport assay. Continue this until all the assays you want to run are running. The duration of the assay is determined by the transport activity and should be determined empirically.
Notes:- We start the assay with 1-2 oocytes more than we want to analyze on the LC-MS as sometimes 1-2 oocytes are lost during the washing steps.
- Transfer the oocytes in only one drop of the Pasteur pipette.
- We start the assay with 1-2 oocytes more than we want to analyze on the LC-MS as sometimes 1-2 oocytes are lost during the washing steps.

Figure 2. Process of transferring oocytes from one well to another using a Pasteur pipette. Please note how the oocytes are allowed to settle in the tip and are expelled in a single drop.
- With a < 2 µl pipette, take out 1 µl of each assay media (Figure 1C) and add it to the Eppendorf tube for the media sample (in this example tube 1).
- Stop the assay by adding 1 ml cold (4 °C) Kulori media (pH 7.4) to the assay well and immediately transfer the oocytes to the first Petri dish using a Pasteur pipette. Make sure to transfer oocytes in only one drop from the pipette and empty the rest of the solution in the pipette into the waste. Subsequently, and in the same way move the oocytes to Petri dish two and then Petri dish three to wash away any external substrate. This washing procedure effectively dilutes the substrate in the external uptake media to below detection levels.
Note: Each time make sure to only transfer the oocytes with one drop of Kulori media.
- Transfer one oocyte to each of the 1.5 ml Eppendorf tubes numbered 2-6 and carefully remove excess wash media with a 100 µl pipette from each tube.
Note: Removal of excess wash media is a key step. Complete removal ensures low variability between replicates.
- Add 50 μl of 50% MeOH (with an appropriate internal standard. For glucosinolate transport assays, we use 1,250 nM of the glucosinolate sinigrin as it is commercially available) to the five oocyte samples and the media sample. Immediately homogenate the oocytes using a 100 µl pipette.
Note: Adding MeOH and waiting will result in oocytes that cannot be homogenated due to the dehydration by MeOH. It is therefore important that the homogenization is carried out immediately.
- Leave the homogenate for two hours at -20 °C and then centrifuge the samples at 20,000 x g for 15 min at 4 °C to precipitate proteins. Transfer 40 µl of the supernatant to the corresponding tube in the second set of numbered tubes and dilute with 60 μl H2O.
- Filter the diluted samples through a 0.22 μm PVDF based filter plate (EMD Millipore) and subsequently analyze by analytical LC-MS.
- To start the first transport assay, preincubate 6-7 oocytes expressing AtGTR1 in the preincubation well containing Kulori media (pH 5.6) for 5 min (Figure 1B). Subsequently, use a Pasteur pipette to transfer oocytes to the assay well containing Kulori media (pH 5.6) with substrate (Figure 1C) and incubate for 2-180 min at room temperature. Make sure to transfer oocytes in only one drop from the pipette (see Figure 2). Wait three minutes and start the next transport assay. Continue this until all the assays you want to run are running. The duration of the assay is determined by the transport activity and should be determined empirically.
- LC-MS analysis of glucosinolates and cyanogenic glucosides
- Analysis can be performed by any type of UHPLC coupled to a Triple Quadrupole mass spectrometer. Separation of analytes is routinely done by reverse phase liquid chromatography using a C18-type column using MilliQ-grade water with 0.05% formic acid and acetonitrile with 0.05% formic acid as gradient solvents. Electrospray ionization (ESI) is then followed by detection by the MS using Multi Reaction Monitoring (MRM) which allows for detection of analytes to very low concentrations depending on ionization efficiency and other instrument parameters. Typically, analytes such as glucosinolates have a lower limit of detection (LLOD) around 5-10 nM (approx. 5-10 fmol on column) (Crocoll et al., 2016) while cyanogenic glucosides have a LLOD of around 200-250 nM (approx. 200-250 fmol on column). The lower limit of quantification (LLOQ) is around 20-50 nM and 400-500 nM for glucosinolates and cyanogenic glycosides, respectively.
- Here, chromatography was performed on an Advance UHPLC system (Bruker, Bremen, Germany). Separation was achieved on a Kinetex 1.7u XB-C18 column (100 x 2.1 mm, 1.7 μm, 100 Å, Phenomenex, Torrance, CA, USA). Formic acid (0.05%) in water and acetonitrile (supplied with 0.05% formic acid) were employed as mobile phases A and B, respectively. The elution profile was: 0-0.2 min, 2% B; 0.2-1.8 min, 2-30% B; 1.8-2.5 min 30-100% B, 2.5-2.8 min 100% B; 2.8-2.9 min 100-2% B and 2.9-4.0 min 2% B. The mobile phase flow rate was 400 μl/min. The column temperature was maintained at 40 °C. The liquid chromatography was coupled to an EVOQ Elite Triple Quadrupole mass spectrometer (Bruker, Bremen, Germany) equipped with an electrospray ion source (ESI) operated in combined positive and negative ionization mode. The instrument parameters were optimized by infusion experiments with pure standards. The ion spray voltage was maintained at 5,000 V or -4,000 V for cyanogenic glucoside and glucosinolate analysis, respectively. Cone temperature was set to 300 °C and cone gas to 20 psi. Heated probe temperature was set to 180 °C and probe gas flow to 50 psi. Nebulizing gas was set to 60 psi and collision gas to 1.6 mTorr. Nitrogen was used as probe and nebulizing gas and argon as collision gas. Active exhaust was constantly on. Multiple Reaction Monitoring (MRM) was used to monitor analyte parent ion → product ion transitions: MRM transitions were chosen based on direct infusion experiments. Detailed values for mass transitions can be found in supplemental Table S3 of Jørgensen et al. (2017). Both Q1 and Q3 quadrupoles were maintained at unit resolution. Bruker MS Workstation software (Version 8.2, Bruker, Bremen, Germany) was used for data acquisition and processing. Linearity in ionization efficiencies was verified by analyzing dilution series of standard mixtures. Quantification of all compounds was achieved by use of sinigrin as an internal standard.
- Analysis can be performed by any type of UHPLC coupled to a Triple Quadrupole mass spectrometer. Separation of analytes is routinely done by reverse phase liquid chromatography using a C18-type column using MilliQ-grade water with 0.05% formic acid and acetonitrile with 0.05% formic acid as gradient solvents. Electrospray ionization (ESI) is then followed by detection by the MS using Multi Reaction Monitoring (MRM) which allows for detection of analytes to very low concentrations depending on ionization efficiency and other instrument parameters. Typically, analytes such as glucosinolates have a lower limit of detection (LLOD) around 5-10 nM (approx. 5-10 fmol on column) (Crocoll et al., 2016) while cyanogenic glucosides have a LLOD of around 200-250 nM (approx. 200-250 fmol on column). The lower limit of quantification (LLOQ) is around 20-50 nM and 400-500 nM for glucosinolates and cyanogenic glycosides, respectively.
- LC-MS analysis–preparation of standards
The LC-MS analysis parameters are highly dependent on the type of equipment available, the setup of the LC-MS system and the compound to be analyzed. It is therefore important to consult the person running the LC-MS equipment prior to starting assays.
We utilize an internal standard (e.g., sinigrin) and an external standard curve to (semi)quantitatively measure the amount of glucosinolates that is taken up into oocytes during the assay. Based on the external standard curve we can calculate a response factor that we can then use to calculate a sample’s substrate concentration. Using an internal standard for analysis has several advantages over only using an external standard curve, e.g., correction for handling during extraction, correction for technical variation during LC-MS acquisition and it does not require running an external standard curve every single time which reduces sample number and running costs (especially when considering triple injection of a standard curve with 10-12 concentrations covering the linear range of detection). The linear range of modern mass spectrometers often covers 4-5 orders of magnitude (e.g., from as low as 1 nM to up 100 µM). The linearity should always be checked as some analytes might show a non-linear behavior or the linear range is reduced.- Prepare your standard dilution series in 20% MeOH (same as samples to be analyzed). The range of your standard dilution series should be determined empirically based on your substrates ionization efficiency and the transporter’s activity. In our case, we prepare a dilution series from 1 nM to 20,000 nM sinigrin in 20% MeOH.
Note: How to empirically determine the correct dilution series range? Perform an uptake experiment and as default use a dilution series from 1 nM to 20,000 nM internal standard. If your sample concentrations are not within the standards linear range, you should increase the range of your dilution series or dilute your sample appropriately.
- Prepare 11 LC-MS vials (one per standard curve concentration) and add 100 µl of the appropriate standard curve solution in each.
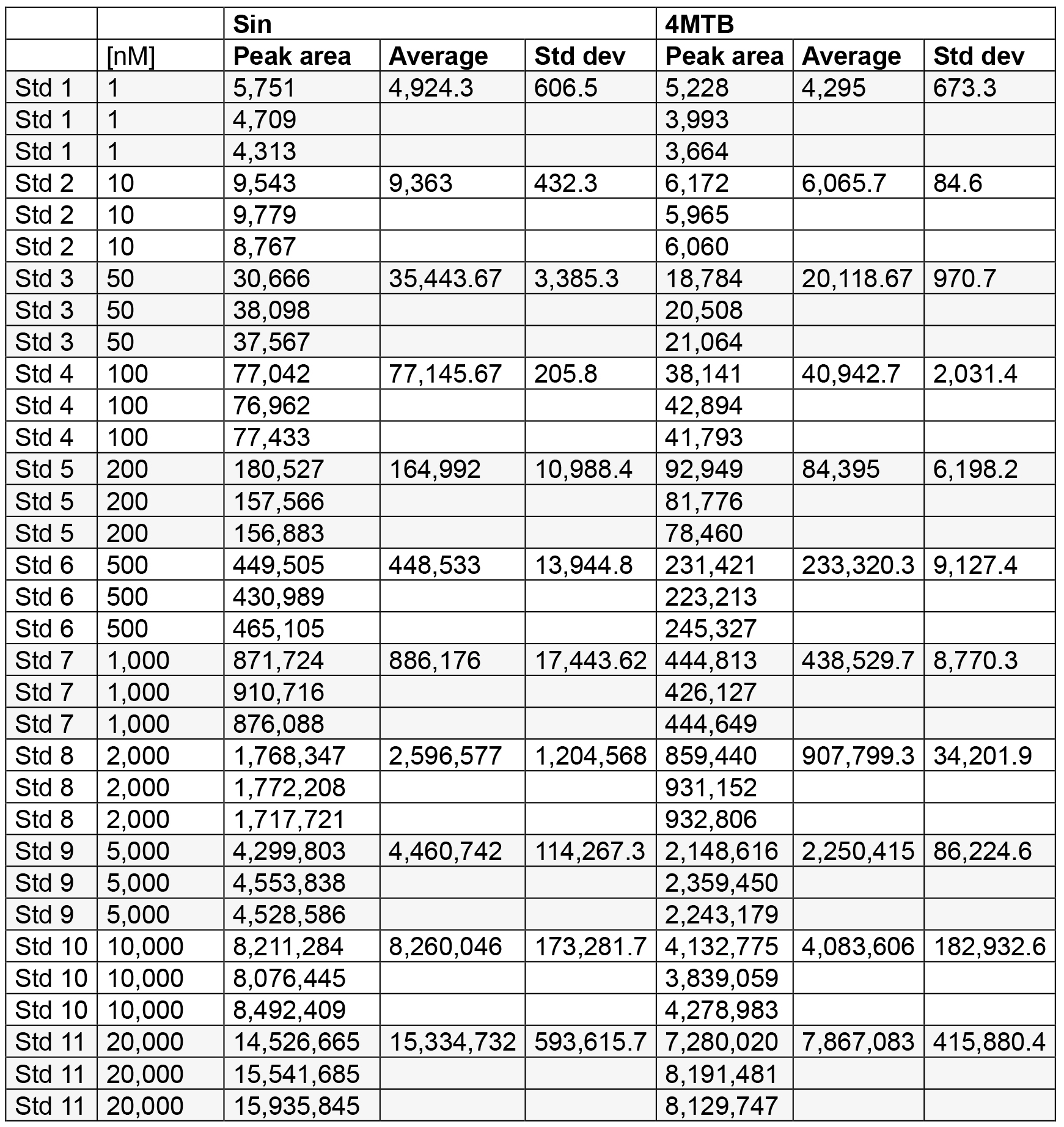
- Standard curve samples are measured by LC-MS in triplicate and an average is calculated (see Table 2).
Table 2. Standard curve measurements
- We plot the analyte concentration (sinigrin and 4MTB in this example) as a function of the signal intensity from the LC-MS and calculate the linear equation to describe the relationship between these two values (see Figure 3) (Crocoll et al., 2016).
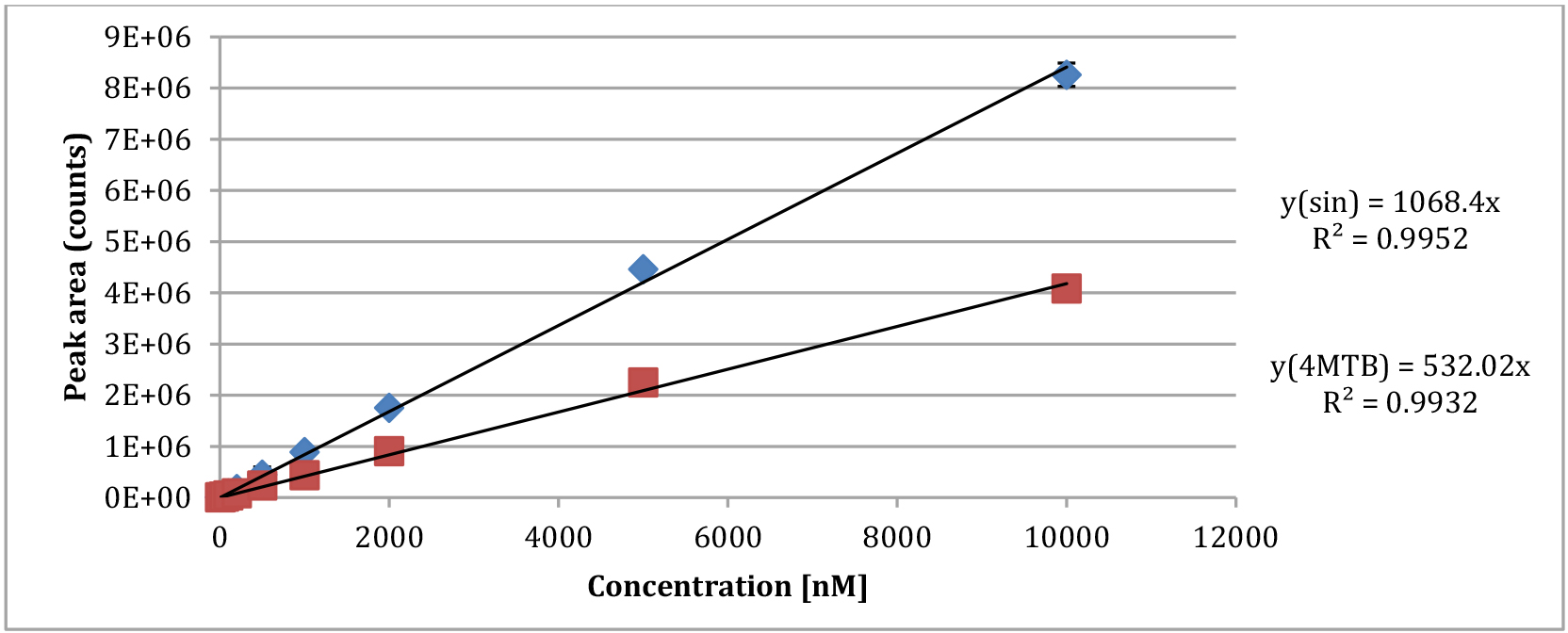
Figure 3. Plot of the standard concentrations relative to the peak area measured by the mass spectrometer
We calculate the response factor (RF) between our internal standard with known concentration and our substrate (4MTB in this example) by dividing the slope of the internal standard with the slope of the substrate.
Note: RF values can NOT be transferred between instruments as the response of each analyte depends on instrument settings for source temperature, ionization energy, collision gas, collision energy and other settings that also can be specific to instruments from different vendors (Crocoll et al., 2016).

The RF value is used to quantify the amount of substrate taken up during our transport assays.
- Prepare your standard dilution series in 20% MeOH (same as samples to be analyzed). The range of your standard dilution series should be determined empirically based on your substrates ionization efficiency and the transporter’s activity. In our case, we prepare a dilution series from 1 nM to 20,000 nM sinigrin in 20% MeOH.
Data analysis
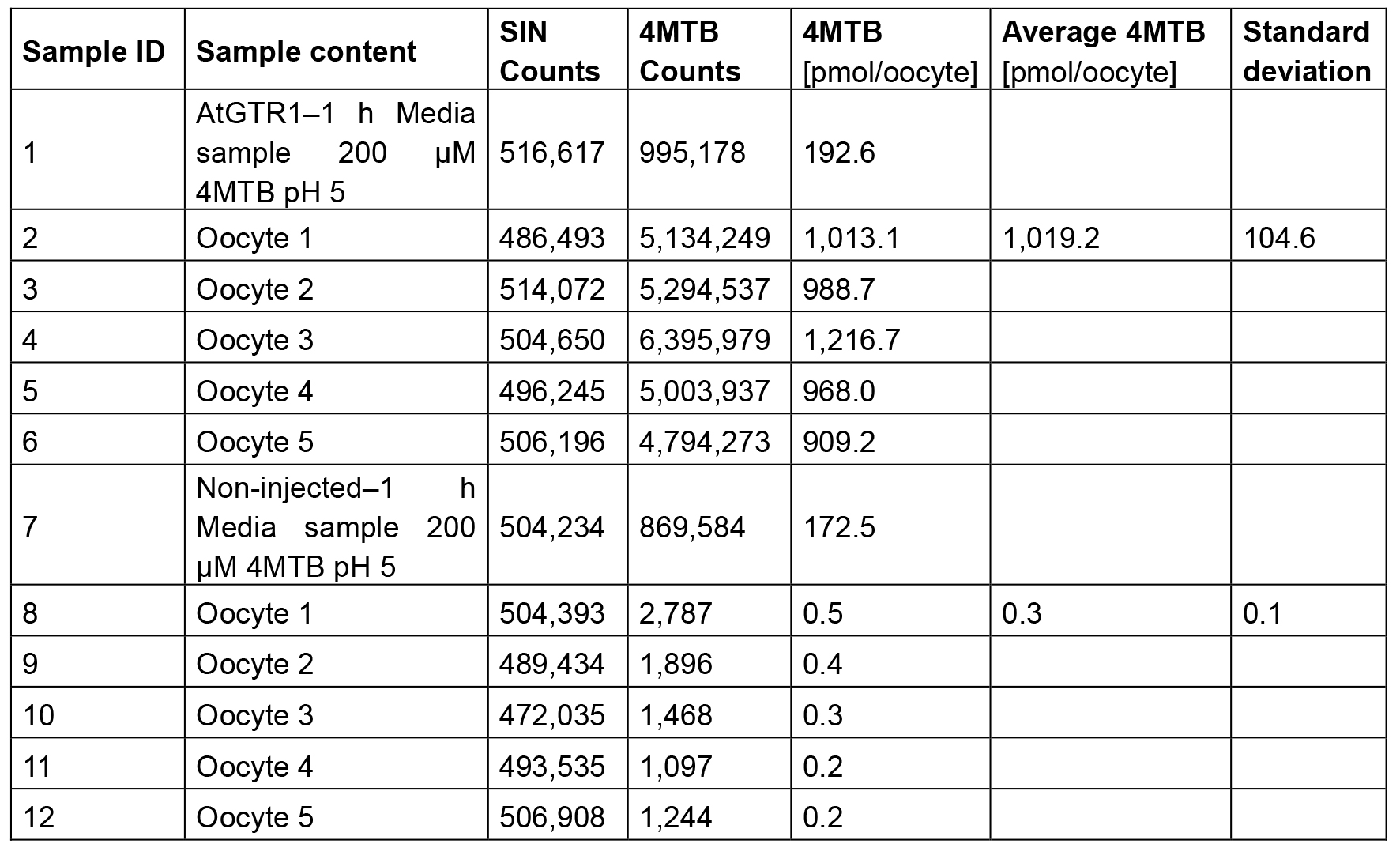
Upon completion of the LC-MS analysis, we calculate the amount of transported substrate into the oocytes. To calculate the number of mol substrate transported per oocyte we multiply the area of the substrate with the amount of internal standard added in the sample and multiplied this with the response factor we calculated in step D4. This value is divided by the area of the internal standard (Equation 2, see Table 3 and Figure 4 for example data).
![]()
The 50% MeOH solution in which we bust oocytes contains 1,250 nM sinigrin as an internal standard. Consequently, we have 50 picomole internal standard in the analyte.
![]()
Table 3. Example of data analysis
Note: These values can be plotted in a bar graph to visually compare uptake by AtGTR1-expressing oocytes and non-expressing control oocytes.
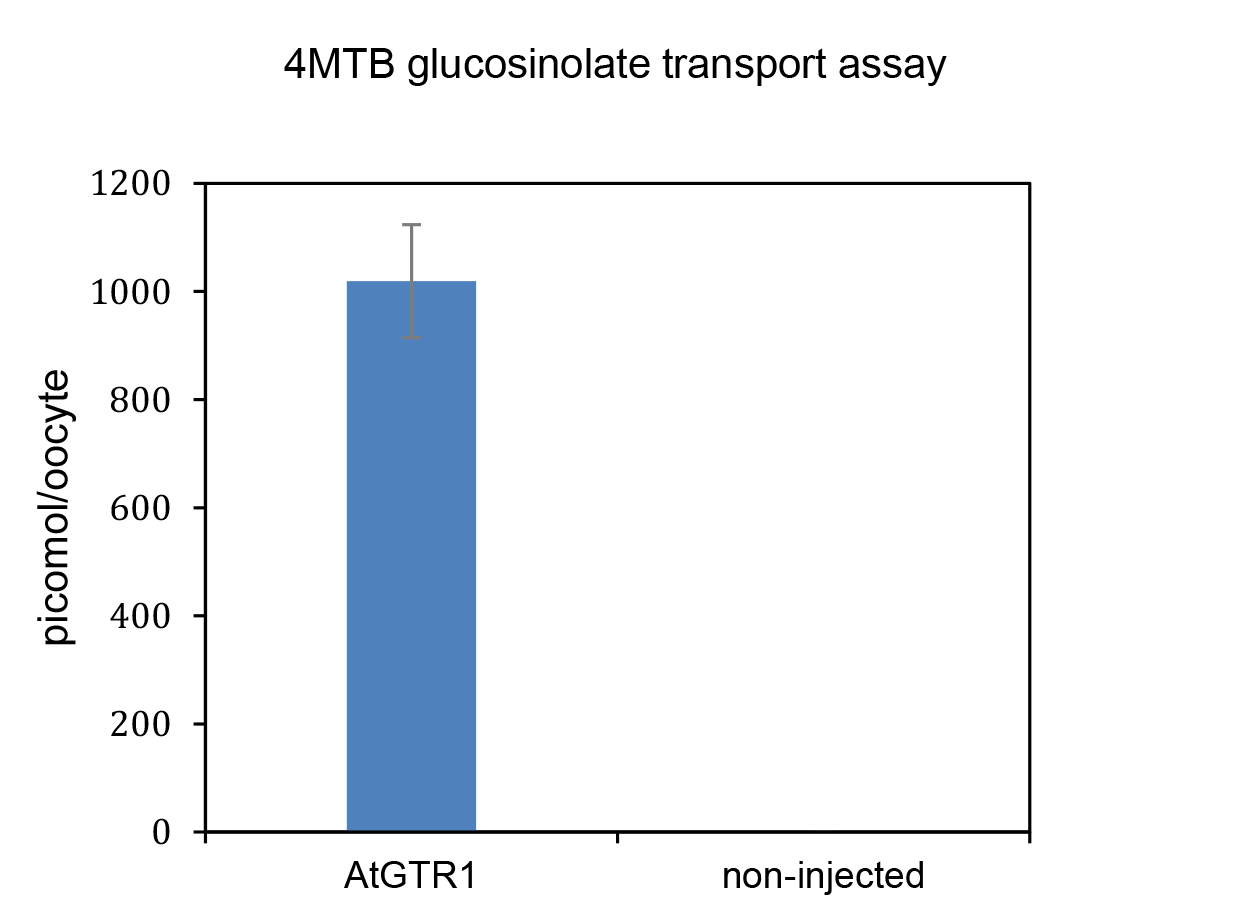
Figure 4. Transport assay with glucosinolate transporter AtGTR1 and non-injected control oocytes
Recipes
- Kulori media (pH 7.4)
90 mM NaCl
1 mM KCl
1 mM MgCl2
1 mM CaCl2
10 mM HEPES
Adjust to pH 7.4 with Tris
- Kulori media (pH 5.6)
90 mM NaCl
1 mM KCl
1 mM MgCl2
1 mM CaCl2
10 mM MES
Adjust to pH 5.6 with Tris
Acknowledgments
We thank Meike Burow and Bo Larsen for their help with the initial LCMS method development for glucosinolate detection from oocyte uptake assays. Morten Egevang Jørgensen is supported by a grant from the Danish Council for Independent Research: DFF–6108-00122. BAH and CC were funded by DNRF99 grant from the Danish National Research Foundation. HHN was funded by DNRF99 grant from the Danish National Research Foundation and by Innovation Fund Denmark J.nr.: 76-2014-3.
References
- Boorer, K. J., Forde, B. G., Leigh, R. A. and Miller, A. J. (1992). Functional expression of a plant plasma membrane transporter in Xenopus oocytes. FEBS Lett 302(2): 166-168.
- Calamita, G., Bishai, W. R., Preston, G. M., Guggino, W. B. and Agre, P. (1995). Molecular cloning and characterization of AqpZ, a water channel from Escherichia coli. J Biol Chem 270(49): 29063-29066.
- Crocoll, C., Halkier, B. A. and Burow, M. (2016). Analysis and quantification of glucosinolates. Curr Protoc Plant Biol 1: 385-409.
- Jørgensen, M. E., Nour-Eldin, H. H. and Halkier, B. A. (2016). A Western blot protocol for detection of proteins heterologously expressed in Xenopus laevis oocytes. Methods Mol Biol 1405: 99-107.
- Jørgensen, M. E. Xu, D., Crocoll, C., Ramírez, D., Motawia, M. S., Olsen, C. E., Nour-Eldin, H. H. and Halkier, B. A. (2017). Origin and evolution of transporter substrate specificity within the NPF family. eLife 6: e19466.
- Norholm, M. H., Nour-Eldin, H. H., Brodersen, P., Mundy, J. and Halkier, B. A. (2006). Expression of the Arabidopsis high-affinity hexose transporter STP13 correlates with programmed cell death. FEBS Lett 580(9): 2381-2387.
- Nour-Eldin, H. H., Norholm, M. H. and Halkier, B. A. (2006). Screening for plant transporter function by expressing a normalized Arabidopsis full-length cDNA library in Xenopus oocytes. Plant Methods 2: 17.
- Sigel, E. (1990). Use of Xenopus oocytes for the functional expression of plasma membrane proteins. J Membr Biol 117(3): 201-221.
- Sumikawa, K., Houghton, M., Emtage, J. S., Richards, B. M. and Barnard, E. A. (1981). Active multi-subunit ACh receptor assembled by translation of heterologous mRNA in Xenopus oocytes. Nature 292(5826): 862-864.
- Theodoulou, F. L. and Miller, A. J. (1995). Xenopus oocytes as a heterologous expression system for plant proteins. Mol Biotechnol 3(2): 101-115.
- Wahl, R., Wippel, K., Goos, S., Kamper, J. and Sauer, N. (2010). A novel high-affinity sucrose transporter is required for virulence of the plant pathogen Ustilago maydis. PLoS Biol 8(2): e1000303.
Article Information
Copyright
![]() Jørgensen et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Jørgensen et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Jørgensen, M. E., Crocoll, C., Halkier, B. A. and Nour-Eldin, H. H. (2017). Uptake Assays in Xenopus laevis Oocytes Using Liquid Chromatography-mass Spectrometry to Detect Transport Activity. Bio-protocol 7(20): e2581. DOI: 10.21769/BioProtoc.2581.
- Jørgensen, M. E. Xu, D., Crocoll, C., Ramírez, D., Motawia, M. S., Olsen, C. E., Nour-Eldin, H. H. and Halkier, B. A. (2017). Origin and evolution of transporter substrate specificity within the NPF family. eLife 6: e19466.
Category
Molecular Biology > Protein > Expression
Biochemistry > Protein > Activity
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.