- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Construction of a Single Transcriptional Unit for Expression of Cas9 and Single-guide RNAs for Genome Editing in Plants
Published: Vol 7, Iss 17, Sep 5, 2017 DOI: 10.21769/BioProtoc.2546 Views: 13727
Reviewed by: Rainer MelzerAlberto CarbonellAnonymous reviewer(s)
Original research article
The authors used this protocol in:
Jul 2016

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
The CRISPR (clustered regularly interspaced short palindromic repeats)-associated protein9 (Cas9) is a simple and efficient tool for genome editing in many organisms including plant and crop species. The sgRNAs of the CRISPR/Cas9 system are typically expressed from RNA polymerase III promoters, such as U6 and U3. In many transformation events, more nucleotides will increase the difficulties in plasmid construction and the risk of wrong integration in genome such as base-pair or fragment missing (Gheysen et al., 1990). And also, in many organisms, Pol III promoters have not been well characterized, and heterologous Pol III promoters often perform poorly (Sun et al., 2015). Thus, we have developed a method using single transcriptional unit (STU) CRISPR-Cas9 system to drive the expression of both Cas9 and sgRNAs from a single RNA polymerase II promoter to achieve effective genome editing in plants.
Keywords: CRISPRBackground
The sgRNA of the CRISPR-Cas9 system is mainly promoted by the small nuclear RNA promoters such as U6 and U3. Although it has been tested with prospered efficiency in many cases, it also has some limitations: (1) it is hard to achieve coordinated and/or inducible expression of Cas9 and the sgRNAs; (2) manipulating multiple sgRNAs for multiplexed gene editing can be tedious, requiring multiple Pol III promoters. The traditional RNA polymerase II promoter can’t be used in driving sgRNA expression, extra nucleotides will be added to the 5’- and 3’-ends of gRNA by RNA polymerase II and may interrupt the normal gRNA function. Additionally, RNAs transcribed by RNA polymerase II are exported rapidly into the cytoplasm while nuclear localization is required for the CRISPR-Cas9/gRNA duplex to access the genome editing (Lei et al., 2001). To overcome these obstacles, we use the ribozyme’s self-catalyzed cleavage to release the precise processing mature sgRNA under a RNA polymerase II promoter which drives expression of both Cas9 and sgRNA (named STU CRISPR-Cas9 system, Figure 1). Compared to the traditional small nuclear RNA promoters used in sgRNA expression, our STU CRISPR-Cas9 system has some advantages: (1) it’s shorter and easier in vector construction, and it will increase the transformation efficiency under some circumstances; (2) it only needs extra ribozyme flanking sequence (shorter than any RNA polymerase III promoter we are currently using) for multiple sgRNAs expression;(3) it has shown higher deletion efficiency induced by double sgRNAs. Thus, the STU CRISPR-Cas9 system driven by a single RNA polymerase II promoter can replace the traditional CRISPR-Cas9 system now we are using whether in vivo or in vitro if appropriate promoters are chosen.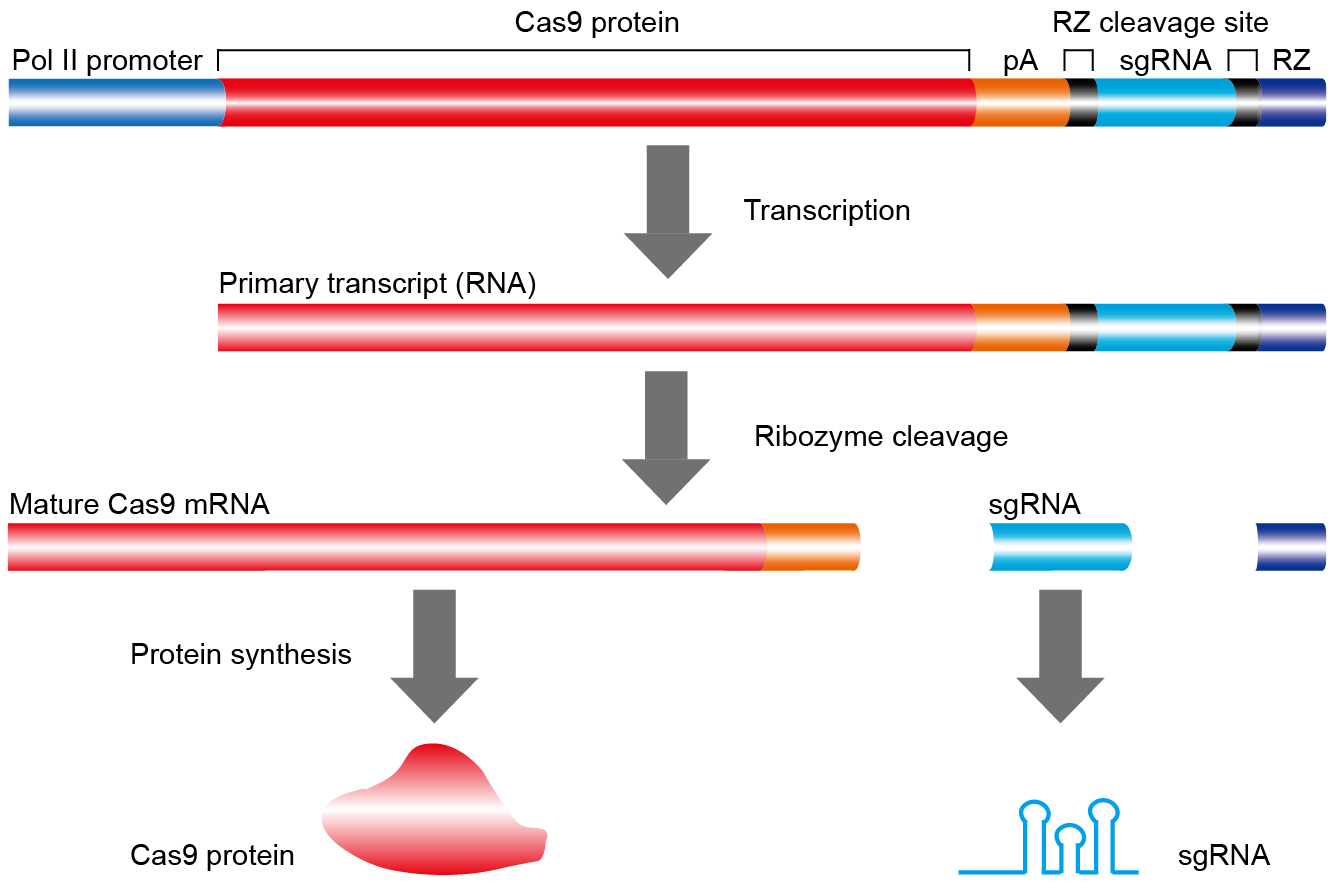
Figure 1. Schematic illustration of the single transcription unit (STU) CRISPR-Cas9 system. Once transcribed by a Pol II promoter, the STU CRISPR-Cas9 primary transcripts will undergo self-cleavage by hammerhead ribozyme (RZ) to release the mature Cas9 mRNA and sgRNA. The Cas9 mRNA is terminated with a synthetic polyA (pA) sequence to facilitate translation, The RZ sequence (in blue) and its target sequence (in black) are illustrated.
Materials and Reagents
- 0.2 ml PCR tubes (Biosharp, catalog number: BS-02-P )
- 1.5 ml Eppendorf tubes (Biosharp, catalog number: BS-15-M )
- Pipette tips (Biosharp, catalog numbers: BS-10-T , BS-200-T , BS-1000-T )
- Competent E. coli DH5α cells (Homemade)
- pTX171 plasmids (Addgene, catalog number: 89258 )
- pTX172 plasmids (Addgene, catalog number: 89259 )
- BsaI (New England Biolabs, catalog number: R0535L )
- Deionized water (sterile)
- Agarose (Biowest, catalog number: 111860 )
- Ethidium bromide (Solarbio Life Scientific, catalog number: E1020 )
- AxyPrepTM DNA Gel Extraction Kit (Corning, Axygen®, catalog number: AP-GX-250 )
- T4 DNA ligase (New England Biolabs, catalog number: M0202L )
- dNTPs mixture (Tiangen Biotech, catalog number: CD117-11 )
- Taq DNA polymerase (Tiangen Biotech, catalog number: ET101-01-02 )
- Q5® High-Fidelity DNA polymerase (New England Biolabs, catalog number: M0491L )
- AxyPrepTM Plasmid Miniprep Kit (Corning, Axygen®, catalog number: AP-MN-P-250 )
- Kanamycin (Solarbio Life Scientific, catalog number: K8020 )
- TAE electrophoresis buffer (see Recipes)
Tris (Solarbio Life Scientific, catalog number: T8060 )
Acetic acid (Kelong)
0.5 M EDTA (Solarbio Life Scientific, catalog number: E1170 )
- LB medium (see Recipes)
Tryptone (Oxoid, catalog number: LP0042 )
Yeast extract (Oxoid, catalog number: LP0021 )
Sodium chloride (NaCl) (Kelong)
Equipment
- Pipettes (Dragon-Lab)
- Heating block (Hangzhou Allsheng Instruments, model: MK-20 )
- Thermal cycler (Thermo Fisher Scientific, Thermo ScientificTM, model: ArktikTM Thermal Cycler )
- Water bath (Yongguangming, model: DZKW-S-4 )
- Microcentrifuge (Eppendorf, model: 5424 )
- DNA electrophoresis apparatus (Bio-Rad Laboratories, model: Mini-Sub® Cell GT Systems )
- NanoDrop (Thermo Fisher Scientific, Thermo ScientificTM, model: NanoDropTM 2000 )
Procedure
- Design sgRNAs to target the genes of interest
- Select appropriate sgRNA targets for the genes of interest using the online sgRNA design tools such as CRIPSR-P v2.0 (http://cbi.hzau.edu.cn/CRISPR2/); CRISPR RGEN tools (http://www.rgenome.net/cas-offinder/); E-CRISP (http://www.e-crisp.org/E-CRISP/designcrispr.html). These are several web-based tools available for sgRNA design. They have mainly the same functions in sgRNA design and Off-target prediction. The main difference is the algorithm of the scoring system.
Notes: sgRNA targets containing a restriction enzyme site at the Cas9 cleavage site would contribute to identify mutant using polymerase chain reaction-restriction endonuclease digestion assay.
- Design and order forward and reverse oligonucleotides for cloning sgRNA into the STU CRISPR-Cas9 expression vector. (1) the forward sgRNA oligonucleotide contains a ‘CGGA’ sequence at the 5’ end followed by 20 bases of sgRNA targets without PAM sites (N20); (2) the reverse sgRNA oligonucleotide contains an ‘AAAC’ at the 5’ end followed by the reverse complement of N20.
For example, if the target site is GTTGGTCTTTGCTCCTGCAGAGG (AGG is PAM), the forward and reverse oligonucleotides should be:
Forward oligonucleotide:5’-CGGAGTTGGTCTTTGCTCCTGCAG-3’
Reverse oligonucleotide:5’-AAACCTGCAGGAGCAAAGACCAAC-3’
- Select appropriate sgRNA targets for the genes of interest using the online sgRNA design tools such as CRIPSR-P v2.0 (http://cbi.hzau.edu.cn/CRISPR2/); CRISPR RGEN tools (http://www.rgenome.net/cas-offinder/); E-CRISP (http://www.e-crisp.org/E-CRISP/designcrispr.html). These are several web-based tools available for sgRNA design. They have mainly the same functions in sgRNA design and Off-target prediction. The main difference is the algorithm of the scoring system.
- Annealing of sgRNA oligos
- Mix 10 µl of the forward and reverse oligos (100 µM) of each sgRNA in separate microtubes.
- Incubate the microtubes at 95 °C for 5 min in a heating block or thermal cycler.
- Allow the microtubes to slowly cool down to room temperature.
- Make a 1:200 dilution of the annealed mixture with deionized water.
- Mix 10 µl of the forward and reverse oligos (100 µM) of each sgRNA in separate microtubes.
- Vector cloning
Two methods could be used to clone sgRNAs into the STU CRISPR-Cas9 expression vector: Cut and ligation or Golden Gate method (Figure 2). These two methods are the same in procedure including BsaI digestion and T4 DNA ligase ligation. The Golden Gate reaction is much easier because the digestion and ligation will process in one PCR tube, and it may save some time.
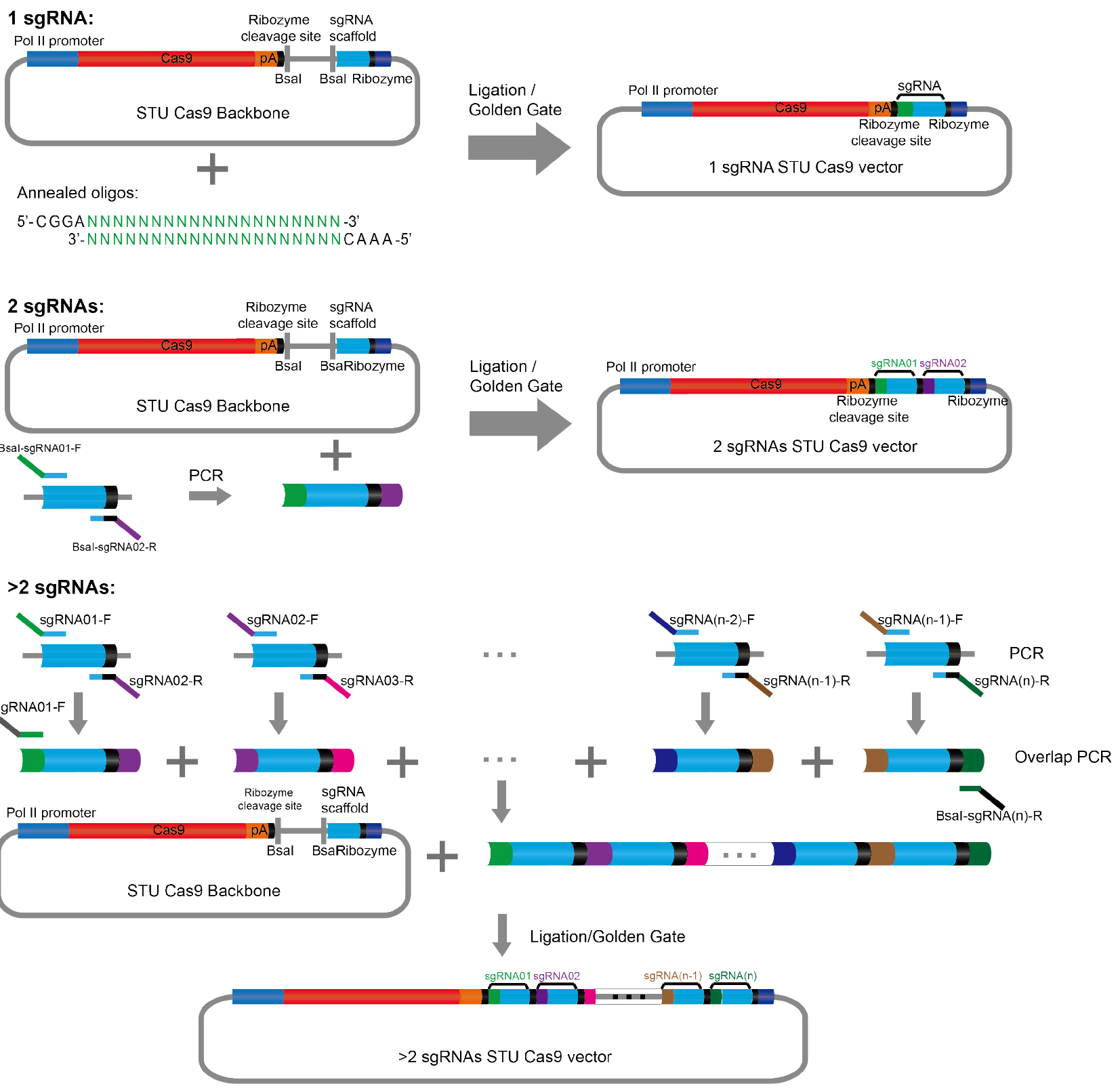
Figure 2. Schematic illustration of the cloning procedure described in the protocol
- Cut and ligation
- Linearize the STU CRISPR-Cas9 plasmid pTX171 or pTX172 with BsaI. Incubate at 37 °C for 2-4 h.
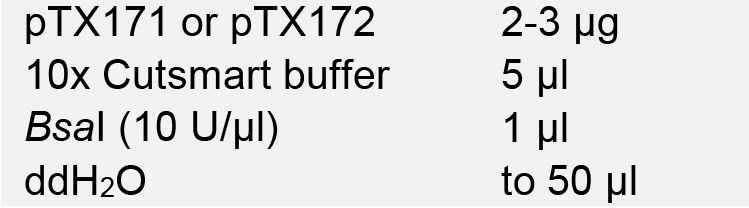
- Load digestion products onto a 1% agarose gel for electrophoresis. Purify the digested vector using the AxyPrepTM DNA Gel Extraction Kit, and quantify DNA concentration using NanoDrop.
- Ligate the diluted annealed oligos into linearized STU CRISPR-Cas9 expression vector, incubate at 16 °C overnight or room temperature for 1-2 h.

- Transform 5 μl of the reaction into 50 μl competent DH5ɑ cells, spread the transformed cells on LB plates supplemented with 50 mg/L kanamycin, and then incubate overnight at 37 °C.
- Verify the positive clones by colony PCR and Sanger sequencing.
- Linearize the STU CRISPR-Cas9 plasmid pTX171 or pTX172 with BsaI. Incubate at 37 °C for 2-4 h.
- Golden Gate method (Make sure annealed sgRNA oligos don’t contain BsaI site)
- Set up a Golden Gate reaction for cloning sgRNAs into the STU Cas9 expression vector.

- Incubate Golden Gate reactions in a thermal cycler using the following program: 10 cycles of 5 min at 37 °C and 10 min at 16 °C, then heat to 37 °C for another 5 min and 80 °C for 10 min.
- Transform 5 μl of the reaction into 50 μl competent DH5α cells, spread the transformed cells on LB plates supplemented with 50 mg/L kanamycin, and then incubate overnight at 37 °C.
- Verify the positive clones by colony PCR and Sanger sequencing. Colony PCR can be performed with the forward sgRNA oligonucleotide (e.g., see step A2) and ZY065-RB: (5’-ttctaataaacgctcttttctct-3’). The expected product size is approximately 230 bp. ZY065-RB can be used for sequencing.
- Set up a Golden Gate reaction for cloning sgRNAs into the STU Cas9 expression vector.
- Cut and ligation
- Two sgRNAs can be cloned into the STU CRISPR-Cas9 expression vector to target two sites simultaneously (Figure 2).
- Design two primers as follows:
BsaI-sgRNA01-F: 5’-CAGGTCTCACGGA-N20-gttttagagctagaaatagcaagttaa-3’
BsaI-sgRNA02-R: 5’-TCGGTCTCCAAAC-N20-tccggtgacaaaagcaccga-3’
GGTCTC is the BsaI recognition sequence;
‘N20’ is same as the sgRNA01 target-specific sequence;
‘N20’ is the reverse complement of the sgRNA02 target-specific sequence;
The lowercase letters are complementary with the STU CRISPR-Cas9 expression vector.
Note: PAGE purified oligos are highly recommended, desalted is also OK.
- Set up a 50 μl PCR reaction to amplify DNA for STU CRISPR-Cas9 expression vector construction.

- Run PCR in a thermal cycler with the following program:
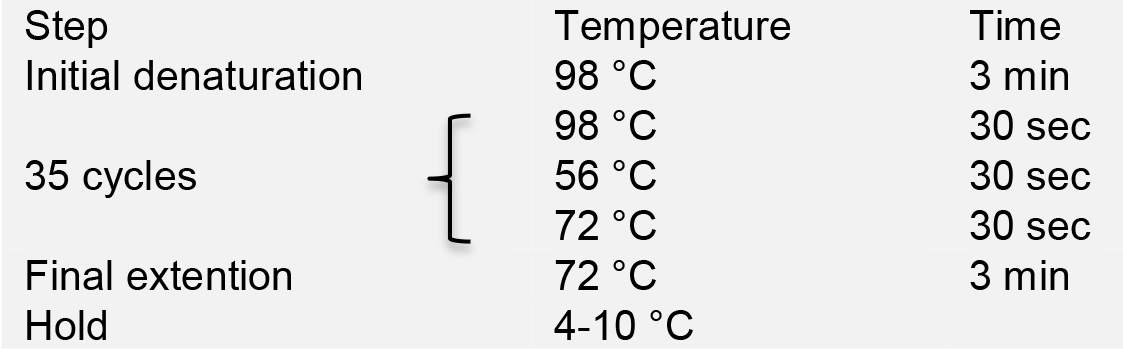
- Load PCR products onto a 1% agarose gel for electrophoresis. Purify the PCR products using the AxyPrepTM DNA Gel Extraction Kit, and quantify DNA concentration using NanoDrop.
- Set up a Golden Gate reaction for cloning two sgRNAs into the STU CRISPR-Cas9 expression vector.

- Incubate Golden Gate reactions in a thermal cycler using the following program: 10 cycles of 5 min at 37 °C and 10 min at 16 °C, then heat to 37 °C for other 5 min and 80 °C for 10 min.
- Transform 5 μl of the reaction products into 50 μl competent DH5α cells, spread the transformed cells on LB plates supplemented with 50 mg/L kanamycin, and then incubate overnight at 37 °C.
- Verify the positive clones by colony PCR and Sanger sequencing.
- Design two primers as follows:
- Our STU CRISPR-Cas9 system has the potential for multiplex sites genome editing. For more than two sites within one STU CRISPR-Cas9 vector, two-round PCR could be performed to clone different sgRNAs into the expression vector (Figure 2).
- Design primers as follows:
BsaI-sgRNA01-F: 5’-CAGGTCTCACGGA-N20-3’
sgRNA01-F: 5’-N20-gttttagagctagaaatagcaagttaa-3’
sgRNA02-F: 5’-N20-gttttagagctagaaatagcaagttaa-3’
sgRNA02-R: 5’-N20-tccggtgacaaaagcaccga-3’
…
sgRNA(n-1)-F: 5’-N20-gttttagagctagaaatagcaagttaa-3’
sgRNA(n-1)-R: 5’-N20-tccggtgacaaaagcaccga-3’
sgRNA(n)-R: 5’-N20-tccggtgacaaaagcaccga-3’
BsaI-sgRNA(n)-R: 5’-TCGGTCTCCAAAC-N20-3’
GGTCTC is the BsaI recognition sequence;
‘N20’ is same as the target-specific sequence;
‘N20’ is the reverse complement of the target-specific sequence;
The different colors represent different target-specific sequences (e.g., N20, N20, N20, N20).
The lowercase letters are complementary with the STU CRISPR-Cas9 expression vector.
- Set up 1st round PCR reactions to amplify sgRNAs fragments with ribozyme cleavage site flanked using primer pairs (sgRNA01-F/sgRNA02-R, sgRNA02-F/sgRNA03-R…sgRNA(n-1)-F/sgRNA(n)-R).

- Run PCR in a thermal cycler with the following program:
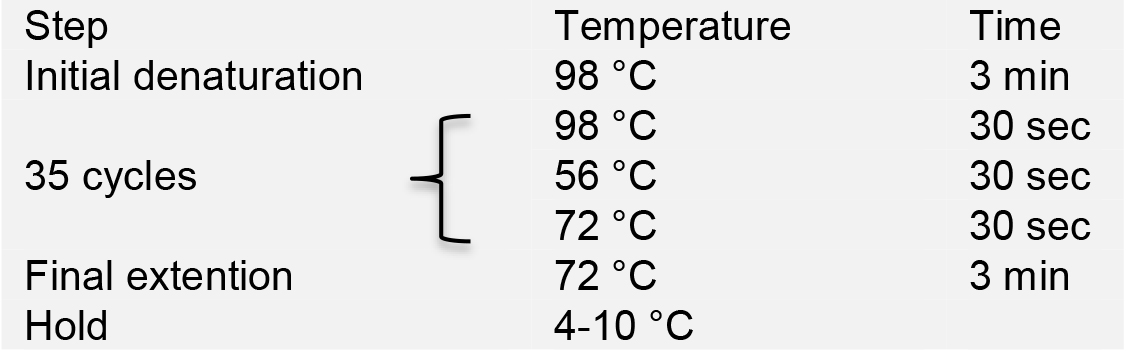
- Load PCR products onto a 1% agarose gel for electrophoresis. Purify the PCR products using the AxyPrepTM DNA Gel Extraction Kit, and quantify DNA concentration using NanoDrop.
- Set up 2nd round PCR reaction to link the different sgRNAs with ribozyme cleavage site into one fragment.

- Run PCR in a thermal cycler with the following program:
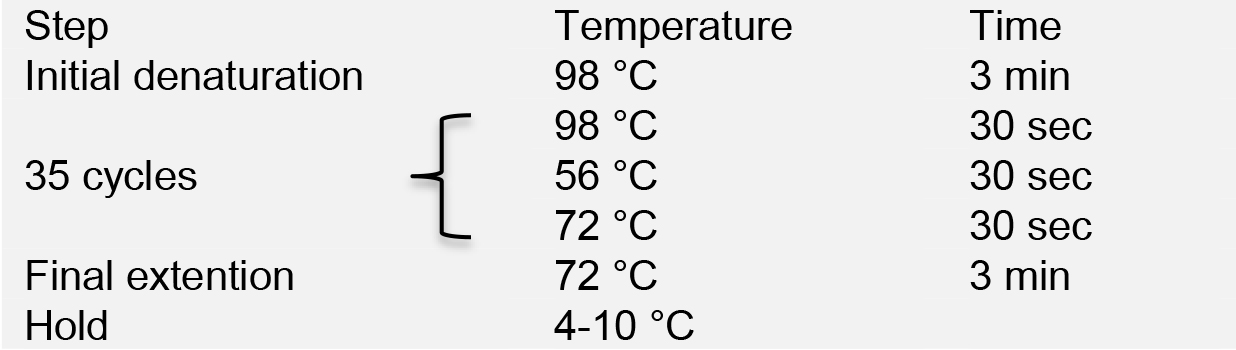
- Load PCR products onto a 1% agarose gel for electrophoresis. Purify the PCR products using the AxyPrepTM DNA Gel Extraction Kit, and quantify DNA concentration using NanoDrop.
- Set up a Golden Gate reaction for cloning multiplex sgRNA Fragment into the STU CRISPR-Cas9 expression vector.

- Incubate Golden Gate reactions in a thermal cycler using the following program: 10 cycles of 5 min at 37 °C and 10 min at 16 °C, then heat to 37 °C for other 5 min and 80 °C for 10 min.
- Transform 5 μl of the reaction products into 50 μl competent DH5α cells, spread the transformed cells on LB plates supplemented with 50 mg/L kanamycin, and then incubate overnight at 37 °C.
- Verify the positive clones by colony PCR and Sanger sequencing.
- Design primers as follows:
Data analysis
Examples of STU CRISPR-Cas9 system application including gene editing and gene deletion with sequencing data in rice, tobacco and Arabidopsis can be found in the original paper (Tang et al., 2016; Link to paper). Additionally, diagrams of the procedure, as well as examples of genome editing and sgRNA multiplex construction, can also be found in the original research paper (Tang et al., 2016).
Notes
- If failed to get colony using Golden Gate method, increase the number of cycles to 15-20 times.
- We tested the STU CRISPR-Cas9 system in several organisms (including rice, tobacco and Arabidopsis) and successfully achieved efficient genome editing.
Recipes
- 50x TAE electrophoresis buffer
242 g/L Tris
57.1 ml/L acetic acid
100 ml/L 0.5 M EDTA (pH 8.0)
- LB medium
10 g/L tryptone
10 g/L NaCl
5 g/L yeast extract
Acknowledgments
Y.Z. was supported by grants from the National Science Foundation of China (31330017 and 31371682), the Sichuan Youth Science and Technology Foundation (2017JQ0005) and the Fundamental Research Funds for the Central Universities (ZYGX2016J119 and ZYGX2016J122). This protocol is developed based on our previous study published in Molecular Plant (Tang et al., 2016).
References
- Gheysen, G., Herman, L., Breyne, P., Gielen, J., Montagu, M. V. and Depicker, A. (1990). Cloning and sequence analysis of truncated T-DNA inserts from nicotiana tabacum. Gene 94(2): 155-63.
- Lei, E. P., Krebber, H. and Silver, P. A. (2001). Messenger RNAs are recruited for nuclear export during transcription. Genes Dev 15(14): 1771-1782.
- Sun, X., Hu, Z., Chen, R., Jiang, Q., Song, G., Zhang, H. and Xi, Y. (2015). Targeted mutagenesis in soybean using the CRISPR-Cas9 system. Sci Rep 5: 10342.
- Tang, X., Zheng, X., Qi, Y., Zhang, D., Cheng, Y., Tang, A., Voytas, D. F. and Zhang, Y. (2016). A single transcript CRISPR-Cas9 system for efficient genome editing in plants. Mol Plant 9(7): 1088-1091.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Tang, X., Zhong, Z., Zheng, X. and Zhang, Y. (2017). Construction of a Single Transcriptional Unit for Expression of Cas9 and Single-guide RNAs for Genome Editing in Plants. Bio-protocol 7(17): e2546. DOI: 10.21769/BioProtoc.2546.
Category
Plant Science > Plant molecular biology > DNA > Mutagenesis
Molecular Biology > DNA > DNA cloning
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.