- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Separation and Purification of Glycosaminoglycans (GAGs) from Caenorhabditis elegans

Published: Vol 7, Iss 15, Aug 5, 2017 DOI: 10.21769/BioProtoc.2437 Views: 12340
Reviewed by: Peichuan ZhangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The nematode Caenorhabditis elegans is a popular model organism for studies of developmental biology, neurology, ageing and other fields of basic research. Because many developmental processes are regulated by glycosaminoglyans (GAGs) on cell surfaces and in the extracellular matrix, methods to isolate and analyze C. elegans GAGs are needed. Such methods have previously been optimized for other species such as mice and zebrafish. After modifying existing purification protocols, we could recently show that the nematodes also produce chondroitin sulfate, in addition to heparan sulfate, thus challenging the view that only non-sulfated chondroitin was synthesized by C. elegans. We here present our protocol adapted for C. elegans. Since the purification strategy involves separation of non-sulfated and sulfated GAGs, it may also be useful for other applications where this approach could be advantageous.
Keywords: GlycosaminoglycansBackground
Glycosaminoglycans (GAGs) are linear polysaccharide chains of repeating disaccharide units, which are often substituted with sulfate groups. Except for hyaluronan, which is a non-sulfated GAG synthesized at the plasma membrane without being anchored to any protein, all other GAGs are covalently linked to core proteins, thus forming proteoglycans (PGs). The most common GAGs found on PGs are heparan sulfate (HS) and chondroitin sulfate (CS)/dermatan sulfate, containing N-acetyl-glucosamine and N-acetyl-galactosamine, respectively (Zhang, 2010). During their biosynthesis in the Golgi compartment, which is a non-template driven process, they are subject to multiple modifications, including epimerization of glucuronic acid into iduronic acid and sulfation at various positions (Bulow and Hobert, 2006; Zhang, 2010). The sulfation patterns generated are important for the ability of the GAG chains to interact with growth factors and cytokines, which in turn is essential for their ability to influence development and other important physiological processes (Bishop et al., 2007).
In order to analyze their composition, GAGs need to be purified from crude cell or tissue lysates, most commonly achieved by ion exchange chromatography after protease and nuclease digestion (Ledin et al., 2004). Disaccharides, generated by the action of specific GAG lyases, can then be identified using different chromatography methods or mass spectrometry (Shao et al., 2013; Kiselova et al., 2014). We and several other labs have used reversed-phase ion-pairing (RPIP)-HPLC for detection of different types of GAGs in multiple species (Ledin et al., 2004; Lawrence et al., 2008; Yamada et al., 2011; Holmborn et al., 2012).
C. elegans synthesizes HS with modifications similar to those found in mammals and other organisms, but like other ecdysozoa the nematodes do not produce hyaluronan (Yamada et al., 1999; Toyoda et al., 2000; Lawrence et al., 2008; Townley and Bulow, 2011). However, although vast amounts of non-sulfated chondroitin were detected, CS was not previously identified, giving C. elegans a unique position among the ecdysozoa (Yamada et al., 1999; Toyoda et al., 2000).
In our protocol we separated sulfated and non-sulfated GAGs prior to analysis, facilitating detection of CS occurring in much lower quantities than the non-sulfated chondroitin (Dierker et al., 2016). Here, this method is described in detail.
Materials and Reagents
- Pipette tips (SARSTEDT, catalog numbers: 70.1114.100 , 70.760.502 , 70.762.200 )
- Tissue paper
- 15 ml tubes (SARSTEDT, catalog number: 62.554.502 )
- 50 ml tubes (SARSTEDT, catalog number: 62.547.254 )
- 1.5 ml Screwlock tubes (SARSTEDT, catalog number: 72.692.100 )
- Syringes 1 ml (Terumo Medical, catalog number: SS+01T1 )
- Syringes 2 ml (BD, catalog number: 300185 )
- MicrolanceTM 3 18 G needles (VWR, catalog number: 613-3945 )
- MicrolanceTM 3 23 G needles (VWR, catalog number: 613-3923 )
- MicrolanceTM 3 27 G needles (VWR, catalog number: 613-3834 )
- 10 ml round-bottom tubes
- Parafilm
- 0.45 µm filters (for example EMD Millipore, catalog number: HAWP04700 )
- Petri dishes 10 cm (SARSTEDT, catalog number: 82.1473.001 )
- 10 ml Poly-Prep® Chromatography columns (Bio-Rad Laboratories, catalog number: 7311550 )
- DEAE Sephacel (GE Healthcare, catalog number: 17050001 )
- 1.5 ml reaction tubes (SARSTEDT, catalog number: 72.690.001 )
- 2 ml reaction tubes (SARSTEDT, catalog number: 72.695.500 )
- NAP-10 columns (GE Healthcare, catalog number: 17-0854-02 )
- HPLC tubes (VWR, catalog number: 548-0440 )
- Locks (Fisher Scientific, catalog number: 11521434 )
Note: This product has been discontinued. - C. elegans and bacterial strains
Note: All C. elegans strains as well as E. coli strain HB101 were obtained from the Caenorhabditis Genetics Center (CGC) https://cbs.umn.edu/cgc/home. - Sodium hypochlorite (NaClO 6-14% active Cl) (Sigma-Aldrich, catalog number: 13440 )
- Protease XIV (Sigma-Aldrich, catalog number: P5147-5G )
- Benzonase (Merck, catalog number: 70746-3 )
- Heparin lyase I, II, III (IBEX Pharmaceuticals, catalog numbers: 50-010 , 50-011 , 50-012 )
- Quant-iTTM Broad Range Assay Kit (purchased from Molecular Probes)
- Calcium chloride (CaCl2•2H2O) (Merck, catalog number: 1.02382.0500 )
- Potassium hypochlorite (KOH) (Sigma-Aldrich, catalog number: P1767 )
- Potassium hydroxide (KOH) (Merck, catalog number: 1.05021.0250 )
- Magnesium sulfate heptahydrate (MgSO4•7H2O) (Merck, catalog number: 1.05886 )
- Cholesterol (Sigma-Aldrich, catalog number: C8667 )
- Ethanol (SOLVECO, catalog number: 1015 )
- Potassium dihydrogen phosphate (KH2PO4) (Merck, catalog number: 1.04873.1000 )
- di-Potassium hydrogen phosphate trihydrate (K2HPO4•3H2O) (Merck, catalog number: A257899 )
Note: This product has been discontinued. - 1 M K2HPO4
- Sodium chloride (NaCl) (Riedel-de Haën, catalog number: 31439 )
- Agarose (Sigma-Aldrich, catalog number: A9539 )
- Bacto peptone (BD, BactoTM, catalog number: 211677 )
- Bacteriological agar (VWR, catalog number: 84609.0500 )
- LB broth 1.1 G per tablet (Sigma-Aldrich, catalog number: L7275-500TAB )
- Bacto yeast extract (BD, BactoTM, catalog number: 212750 )
- di-Sodium hydrogen phosphate dodecahydrate (Na2HPO4•12H2O) (Merck, catalog number: 1.06579.100 )
- Magnesium chloride hexahydrate (MgCl2•6H2O) (Sigma-Aldrich, catalog number: M2670-500G )
- Triton X-100 (Fisher Scientific, catalog number: BP151-500 )
- Tris ultrapure (AppliChem, catalog number: A1086.1000 )
- 6 N HCl
- Sodium acetate (NaOAc) (Merck, catalog number: 1.06268.1000 )
- Silver nitrate (AgNO3) (Sigma-Aldrich, catalog number: S6506-5G )
- Acetic acid (Merck, catalog number: 1.00063.2500 )
- HEPES (Sigma-Aldrich, catalog number: H4034 )
- Chondroitinase ABC (Amsbio, catalog number: AMS.E1028-10 ; Sigma-Aldrich, catalog number: C3667 )
- Media for C. elegans growth and handling (see Recipes)
- 1 M CaCl2
- 5 N KOH
- 1 M MgSO4
- Cholesterol solution
- 1 M potassium phosphate buffer pH 6.0 (KPO4)
- Rich Nematode Growth medium (RNGM)
- LB agar
- 2x YT medium
- M9 buffer
- Buffers for GAG purification (see Recipes)
- 20% ethanol
- 1 M MgCl2
- Stock solutions
i.10% Triton X-100
ii.5 M NaCl
iii.1 M CaCl2
iv.1 M Tris-HCl - Protease buffer
- DEAE elution buffer
- DEAE wash buffers
- DEAE wash buffer, pH 8
- DEAE wash buffer, pH 4
- 0.1% (w/v) AgNO3
- 5x chondroitinase ABC buffer
- 2x heparinase lyase buffer
Equipment
- Pipettes (for example Eppendorf, model: Research® plus )
- 125 ml Erlenmeyer flask (autoclaved)
- Rubber bulb
- 10 ml glass pipettes (autoclaved, plugged)
- Centrifuge
- Incubator for C. elegans (Lovibond Thermostatic Cabinet)
- Stereo microscope (Nikon Instruments, model: SMZ745 , eyelens: C-W10xB/22, magnification 2.5x to 50x)
- Hybridization oven (Hybaid Shake ‘n’ Stack)
- 37 °C incubator (Heraeus Instruments)
- SpeedVac (Labconco Freezer Dry System connected to Savant SpeedVac concentrator or Savant Instruments, model: SC11OA SpeedVac® Plus )
- HPLC system
- Merck Hitachi FL Detector L-7485 (Hitachi, model: Model L-7485 )
- Interface D-7000 (Hitachi, model: Model D-7000 )
- Autosampler L-7200 (Hitachi, model: Model L-7200 )
- Pump L-7100 (Hitachi, model: Model L-7100 )
- Column Oven L-7350 (Hitachi, model: Model L-7350 )
- Reagent pumps: Shimadzu LC-10AD (Shimadzu, model: LC-10AD ); Column: Phenomenex Luna 5u C18(2) 100A (Shimadzu)
Software
- D-7000 HPLC System Manager (HSM) software version 4.1
- GraphPad Prism version 5.0b for Mac OS X (GraphPad Software, San Diego California USA, www.graphpad.com
Procedure
A flowchart of the complete procedure is shown in Figure 1.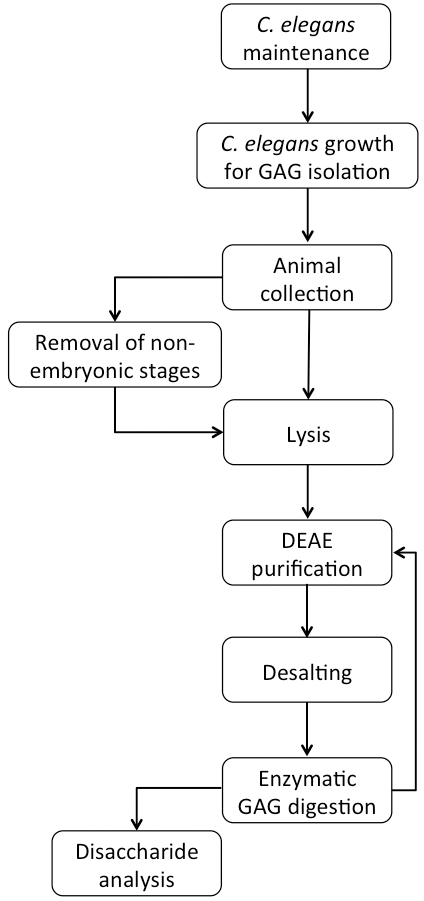
Figure 1. Flowchart of C. elegans growth and GAG purification
Note: Unless mentioned otherwise, all work was performed on a lab bench at room temperature under non-sterile conditions. Handling of C. elegans plates was performed close to an ethanol burner and all pipettes, pipette tips and bottles were quickly heat-sterilized by contact with the ethanol flame. All media and buffer compositions are described under ‘Recipes’ and are not mentioned in the text.
- Growth of C. elegans for GAG analysis
To get large amounts of material for GAG analysis, worms were grown on rich nematode growth medium (RNGM) plates (see Recipes) (10-20 plates, 10 cm diameter) seeded with E. coli HB101. E. coli HB101 were chosen because they are easy for the worms to digest and allow recovery of many worms from the plates:- Streak out HB101 bacteria from a glycerol stock on a fresh LB plate (see Recipes). Grow the bacteria over night at 37 °C.
- Inoculate 50 ml 2x YT medium (see Recipes) with one bacterial colony. Shake overnight (app. 16 h) at 200 rpm in a 125 ml Erlenmeyer flask at 37 °C.
- Prepare 10 cm RNGM plates. Let them dry overnight.
- Next day: Seed plates with a continuous layer (app. 600 µl of bacterial culture, outlined in black in Figure 2) of HB101. Dry the plates overnight on the bench and store until needed at 4 °C in a sealed container containing tissue paper.
Figure 2. Outline of a continuous bacterial layer - Transfer mixed stages of C. elegans to RNGM plates (10 plates per strain for RPIP-HPLC, 20 plates for LC-MS/MS or experiments that require multiple down-stream processing steps) by cutting chunks of C. elegans maintenance plates (prepared according to procedures described here: http://www.wormbook.org/chapters/www_strainmaintain/strainmaintain.html#d0e164), which are placed facedown onto the RNGM plates.
- Grow the animals until the plates contain many worms but make sure that they are not yet starved (i.e., confirm that the plates still have plenty of bacterial food, and that they do not have many dauers, developmentally arrested animals that are often seen under harsh growth conditions, like starvation, heat stress and high population density, etc.). Depending on the strain and the number of transferred animals this normally takes three to seven days.
- Separate plates inoculated with the same strain (10-20 plates, depending on experiment) into piles of five. Collect the worms by pipetting 10 ml M9 buffer (see Recipes) onto the first plate (use a rubber bulb and 10 ml autoclaved glass pipettes). Rinse the plate three times with the same solution and detach worms mechanically by carefully scraping the agar surface with the pipette after the second rinse. Pipette the same 10 ml onto the second plate and repeat until the five plates all have been washed with the same buffer solution. Repeat the procedure with 10 ml of fresh M9 buffer, this time starting with the fifth plate, followed by the fourth until all five plates have been washed twice.
- Pool all washing fractions (in 15 or 50 ml tubes, depending on the sample volume) and collect worms by centrifugation for 4 min at 500 x g.
- Remove the supernatant carefully (pipette and rubber bulb) and disperse the worm pellet in 5-10 ml M9 buffer (depending on bacterial density in buffer and sample volume). Repeat centrifugation and washing until no traces of bacteria are visible (normally two to three washing steps are required).
- Remove M9 buffer from the worm pellet and wash with 5-10 ml ddH2O in order to decrease the remaining amount of salt. Repeat centrifugation as above and transfer pelleted worms (normally between 100 and 500 µl) to a 1.5 ml screw-lock tube.
Note: Cut off the tip of the 1 ml pipette tip to be used for transferring the worm pellet. - Centrifuge as above and remove as much supernatant as possible. Rotate tubes in SpeedVac, apply vacuum (make sure that vacuum is establishd within a few minutes) and dry samples overnight.
Note: If dry weight needs to be determined, the weight of empty tubes should be determined before the worms are added. The difference in weight between the tubes containing the dried worms and empty tubes represents the dry weight of the worm cultures.
- HB101 cultures can be stored at 4 °C for up to two weeks.
- Seeded RNGM plates were usually prepared when needed and should not be stored for longer than two weeks.
- When many RNGM plates are inoculated in parallel, it is important to choose agar chunks that contain approximately the same number of worms, so that all plates will be full of worms at the same time.
- When washing RNGM plates with M9, it is important to reverse the order of plates for the second wash to ensure that all plates are washed with sufficiently clean M9 buffer as bacteria are also washed off.
- If many worms are still present on the plates after two washes, use another 10 ml M9 to collect the residual animals of the same genotype/treatment from up to 10 RNGM plates.
- Dried C. elegans samples can be stored at -20 °C for months without affecting GAG isolation.
- Streak out HB101 bacteria from a glycerol stock on a fresh LB plate (see Recipes). Grow the bacteria over night at 37 °C.
- Isolation of C. elegans eggs for GAG analysis
When GAG analysis was performed on C. elegans embryonic stages, worms were grown as mixed stages (i.e., non-synchronized, thus containing embryonic, larval and adult stages, see http://wormatlas.org/hermaphrodite/introduction/Introframeset.html) on RNGM plates as described above and collected in M9 buffer as described.
Note: The third washing step described in Note e of section A is strongly recommended since eggs tend to stick to the plates.- Pellet worms by centrifugation at 4 °C for 4 min at 2,000 x g and remove the supernatant with a pipette.
- Incubate worm pellets with 3.5 ml ddH2O, 1 ml NaClO and 0.5 ml 5 N KOH until everything but worm eggs is dissolved (approximately 8 min).
Note: Keep the tubes in your hand to increase temperature and rotate them constantly or even mix vigorously. Check the progress of dissolving by regularly inspecting the tubes under a stereo-microscope in order to avoid over-bleaching and killing of the eggs. - Pellet worm eggs by centrifugation at 4 °C for 2 min at 1,300 x g. Remove the supernatant and wash the eggs with 5 ml ddH2O followed by centrifugation at 4 °C for 1 min at 2,000 x g. Repeat the washing step twice.
Note: These conditions might be too harsh to revive larvae from the eggs as they are optimized for collecting eggs that would be homogenized soon after. - Transfer eggs to a screw-lock tube, centrifuge as above and dry under vacuum as described in step A11.
- Pellet worms by centrifugation at 4 °C for 4 min at 2,000 x g and remove the supernatant with a pipette.
- Sample lysis
Sample lysis and purification of GAGs using anion exchange chromatography were adapted from previously described methods (Ledin et al., 2004; Dagalv et al., 2011). If samples had been frozen, they were briefly thawed at room temperature (up to 15 min).- Add 80% of the required final volume (1 ml protease buffer [see Recipes] per 30 mg dry material) and boil the samples for 10 min at 96-100 °C, followed by a brief centrifugation to collect all liquid at the bottom of the tube.
- Pass samples consecutively through needles of decreasing diameters (see Materials and Methods). Use 1 or 2 ml syringes depending on the sample volume. For mixed stages of worms, start lysis with an 18 G needle (10 times passing up and down, carefully avoiding any overload or blockage of the needle), followed by a 23 G needle (same procedure as for 18 G). For worm eggs, it is sufficient to start with a 23 G needle. If possible, pass samples also through a 27 G needle.
Note: 27 G needles tend to clog quickly so use with great care! Use the same syringe for all steps and empty needles as much as possible to avoid loss of material. - Weigh Protease type XIV powder and dissolve in protease buffer to give a final concentration of 0.8 mg/ml. For example, if the final volume is 1 ml, 0.8 mg Protease type XIV is dissolved in 0.2 ml protease buffer and added to the 0.8 ml of homogenized sample.
- Incubate samples at 55 °C for around 24 h with constant rotation in a hybridization oven.
Note: Screw-lock tubes are essential at this step as regular reaction tubes might not remain tight. - Heat-inactivate the protease by a 10 min incubation at 96-100 °C. Centrifuge the samples for 10 min at maximum speed (16,060 x g).
- If DNA concentration will be determined, keep 4 µl of each sample for this purpose.
Note: Can be performed immediately or after storage at -20 °C. - Add 1 M MgCl2 to a final concentration of 2 mM MgCl2 and add 38 U (one unit digests 37 µg DNA in 30 min) Benzonase per ml lysate. Incubate samples overnight (normally 16-18 h) at 37 °C.
Note: If many samples are handled in parallel, a master mix of 1 M MgCl2 and Benzonase is useful. - Inactivate Benzonase followed by centrifugation as described above for Protease inactivation.
- Add 80% of the required final volume (1 ml protease buffer [see Recipes] per 30 mg dry material) and boil the samples for 10 min at 96-100 °C, followed by a brief centrifugation to collect all liquid at the bottom of the tube.
- Ion exchange chromatography for CS purification
Non-sulfated chondroitin (Chn) is highly abundant in C. elegans and may obscure the isolation of sulfated GAGs. The protocol presented below has been optimized for separation of the sulfated GAGs HS and CS (Dierker et al., 2016). Details about incubation times and temperature can be found in Figure 3 and the salt concentration of the buffers used during the different steps is shown in Figure 4.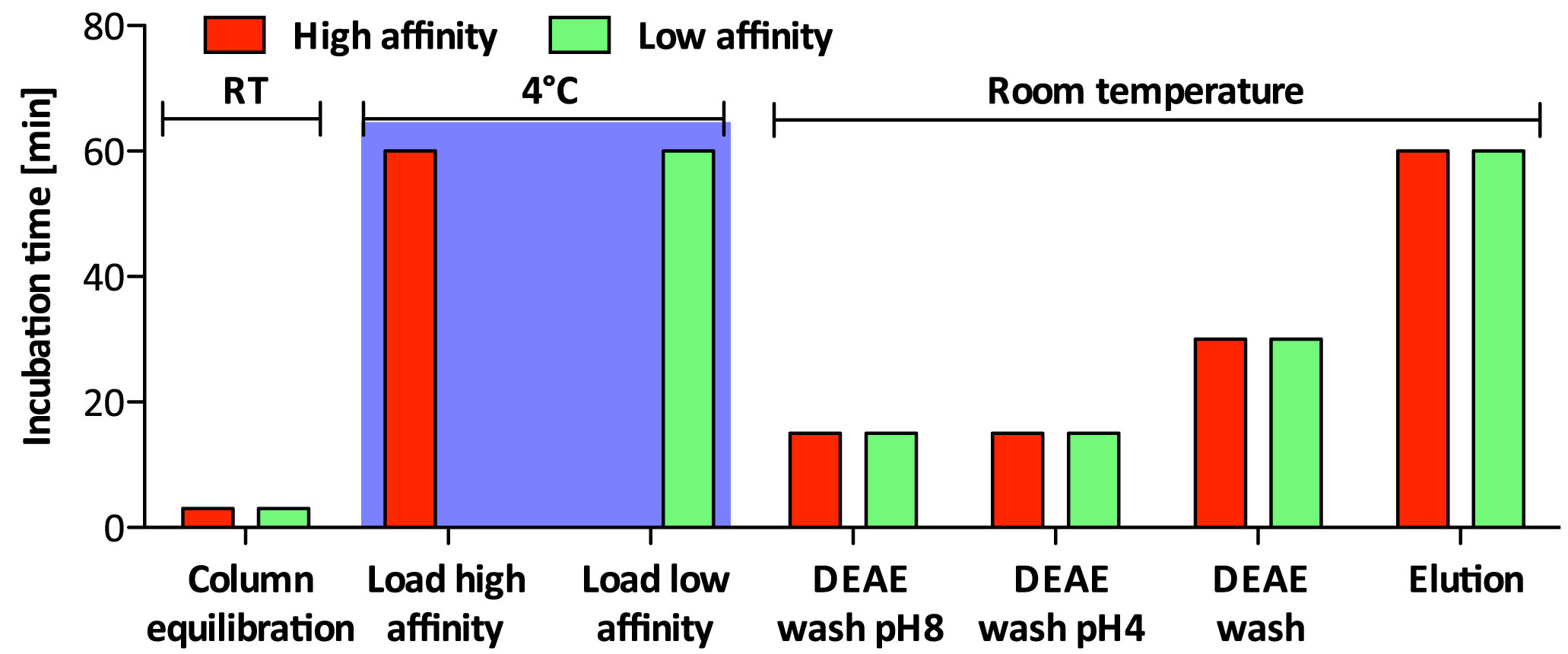
Figure 3. Incubation times and incubation temperature for ion exchange chromatography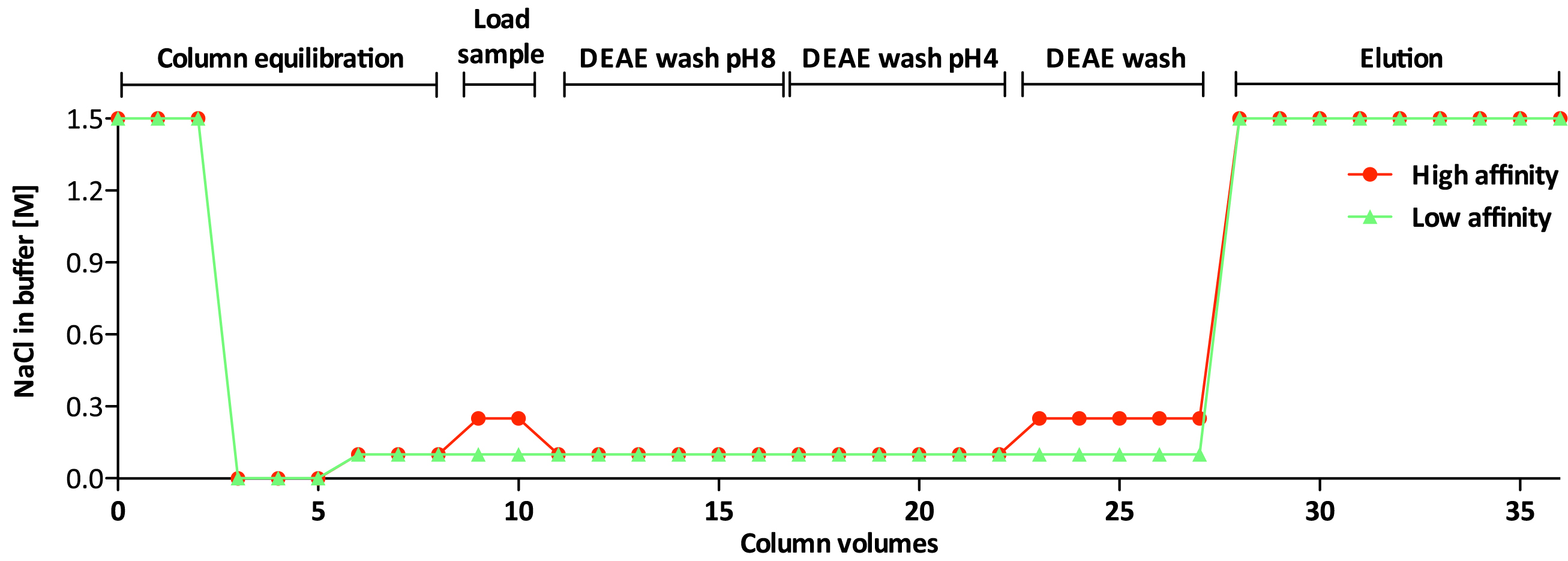
Figure 4. NaCl concentration in high affinity and low affinity GAG-fractions- Transfer sufficient DEAE-Sephacel slurry to 10 ml Poly-Prep® Chromatography columns for bed volumes of 600 µl (Ledin et al., 2004) (see Figure 5).
Note: Slurry can be transferred with a spatula, by pipetting or by pouring it into the column. To maintain the resin in the best state for long-term storage, it is recommended that the slurry is mixed well and that plastic pipette tips are cut off before pipetting.
Figure 5. Final bed volume of DEAE columns - Wash the columns with 2 ml (> three column volumes) 1.5 M NaCl, ddH2O and DEAE wash buffer pH 8 (see Recipes), respectively, before loading the sample.
Note: Columns can be prepared in advance but should then be rehydrated before use. - Adjust the final concentration of NaCl in the lysate to 0.25 M using 5 M NaCl.
- Load the lysates to the pre-washed columns (Note: Avoid any precipitate since this might clog the column–repeat centrifugation described in step C5 if samples have been frozen!). Seal the columns with the provided caps (top) and locks (bottom) as shown in Figure 6. Incubate for 1 h at 4 °C on an orbital rocking table (see Figures 2 and 7).

Figure 6. Sealed DEAE column with sample loaded
Figure 7. Sealed column incubated on a rocking table - Remove caps and locks and collect the flow through fractions in 15 ml reaction tubes. Dilute the flow through fraction with ddH2O to give a final concentration of 0.1 M NaCl before loading it to a second column (prepared in parallel with the first one). Incubate as described in step D4 for 1 h.
Note: GAGs binding to the first column, are designated ‘high affinity’ fraction and GAGs binding to the second column are defined as ‘low affinity’ fraction. - Remove caps and locks and allow the samples to pass through columns.
- Wash all columns with 4 ml DEAE wash buffer pH 8, seal as before and incubate the columns for 10-15 min on a rocking plate at room temperature.
- Remove caps and locks and allow the wash buffer to pass through the columns (see Figure 8), followed by washing with 4 ml DEAE wash buffer pH 4 (see Recipes). Seal as before and incubate for 10-15 min on a rocking plate at room temperature.

Figure 8. Allowing the wash buffer to pass through the column - Remove caps and locks and allow the wash buffer to pass through the columns, followed by washing with 3 ml 0.1 M NaCl (low affinity) or 0.25 M NaCl (high affinity). Seal as before and incubate for 15-30 min on a rocking plate at room temperature.
- Remove caps and locks and allow the wash buffer to pass through the columns. Allow the DEAE-resin to drain completely and add 5.4 ml 1.5 M NaCl per column. Seal and incubate on a rocking plate at room temperature for 1 h in order to elute GAGs efficiently.
- Collect eluates in 10 ml round-bottom tubes. Reduce the volumes of the eluates by SpeedVac centrifugation to approximately 1.7 ml.
- Ensure that the bottom of the columns is sealed before applying the new buffer.
- Parafilm can be used to seal the columns, but the provided locks are very tight and easier to handle.
- Overloading the DEAE columns can reduce recovery. We calculated the required bed volume based on the binding capacity of DEAE for highly sulfated heparin to avoid overloading. However, these calculations might need to be adapted for other samples.
- Transfer sufficient DEAE-Sephacel slurry to 10 ml Poly-Prep® Chromatography columns for bed volumes of 600 µl (Ledin et al., 2004) (see Figure 5).
- Desalting of samples
Desalting was performed using NAP-10 columns:- Wash columns with at least six columns volumes of ddH2O, according to the manufacturer’s instructions.
- Determine the volume of the concentrated eluate and load it.
Note: No more than 0.9 ml of sample can be desalted at a time, so load half of the eluate if the volume is more than that. - Add ddH2O after the sample has entered the column (total volume of steps E2 and E3 should be 1 ml).
- Elute GAGs in 1.0-1.3 ml ddH2O (1.4 ml minus the volume used in step E3) into 10 ml round-bottom tubes.
- Wash columns with at least seven column volumes of ddH2O in order to prepare them for the second half of the eluate and repeat desalting procedure.
- Dry samples completely in a SpeedVac centrifuge.
- NAP-10 columns can be reused. Wash columns with at least seven columns volumes of ddH2O and store them in 20% EtOH at room temperature.
- In order to avoid contamination between samples, columns can also be washed with 1 ml 5 M NaCl followed by exhaustive washing with ddH2O in order to remove any remaining high molecular weight contaminants.
- Dried GAGs normally have a ‘fluffy’, white appearance while residual salt is white, and crystalline. Dissolve samples that are suspected to contain salt in 100 µl ddH2O and mix 1 µl of the sample with a drop of 0.1% AgNO3. If a white cloud appears in the solution (similar to or stronger than in a parallel tube with 1 µl 0.1 M NaCl) dissolve sample in 500 µl ddH2O and repeat desalting. Perform this step for all parallel samples to ensure that they are treated in the same way.
- Wash columns with at least six columns volumes of ddH2O, according to the manufacturer’s instructions.
- GAG degradation and HS purification
The heparinases used by us also show a weak CS-degrading activity, which is why it is important to completely remove CS prior to HS digestion and analysis:- Reconstitute desalted samples in ddH2O.
- Add 20 µl of 5x chondroitinase buffer (final volume: 100 µl) (see Recipes) per tube.
- Add approximately 1 mU chondroitinase (CSase) ABC per mg dried starting material.
- Incubate overnight (minimum 16 h) at 37 °C.
- Heat-inactivate CSase ABC at 96-100 °C for 10 min and centrifuge the samples for 10 min at maximum speed.
- If HS analysis is not carried out, transfer the complete samples to HPLC tubes and adjust volumes to 100 µl with ddH2O to compensate for any loss during the incubation time. Perform disaccharide analysis as described under ‘Data analysis’.
- If in addition information about the HS composition will be determined, save 5-20% of the sample to an HPLC tube for CS compositional analysis.
- Adjust the volume of the remaining sample to 500 µl with ddH2O and 5 M NaCl to achieve a final concentration of 0.1 M NaCl.
- Prepare DEAE columns (200 µl bed volume) as described under Procedure D.
- Load samples, seal columns and incubate for 1 h at 4 °C while rocking.
- Wash the columns with 1.2 ml DEAE wash buffer pH 8, 1.2 ml DEAE wash buffer pH 4 and 1 ml 0.1 M NaCl without further incubation.
- Let the wash buffer drain from columns and elute HS by incubating the columns with 1.8 ml 1.5 M NaCl for 1 h at room temperature while rocking.
- Collect eluates in 2 ml reaction tubes and concentrate them in a SpeedVac centrifuge to 600-800 µl.
- Desalt on NAP-10 columns as described under Procedure E.
Note: The amount of HS in the samples is much lower than that of CS, so barely any precipitate is expected after drying. - Reconstitute the desalted samples in 100 µl ddH2O, divide them into 2x 50 µl aliquots and add 49 µl of 2x HS digestion buffer to both of them.
- Add 1 µl heparinase mix (0.4 mU each of heparinase I, heparinase II, and heparinase III) to one aliquot and 1 µl 1x HS digestion buffer (see Recipes) to the other one.
Note: The non-digested sample is used as a background control in the HPLC-run, see ‘Data analysis’). - Incubate all samples overnight (more than 16 h) at 37 °C.
- Heat-inactivate, centrifuge and transfer the samples to HPLC tubes as described under steps F5 and F6.
- HS and CS digestion buffers can be aliquoted and stored at -20 °C to prolong shelf life.
- Other buffers are stored at room temperature.
- CSase ABC (-20 °C) and heparin lyases (-80 °C) are stored in aliquots of 5-20 µl and multiple freezing is avoided.
- Reconstitute desalted samples in ddH2O.
Data analysis
- Reversed-phase ion-pairing HPLC
Conditions for disaccharide separation (generated by either CSase ABC digestion or by cleavage with a mixture of heparin lyase I, II and III) and post-column derivatisation with 2-cyanoacetamide were described by Ledin et al. (2004) and slightly modified by Dagalv et al. (2011).- Use disaccharide standards analyzed in the same sample series to identify and quantify the peaks based on their elution position and peak size using the D-7000 HPLC System Manager (HSM) software version 4.1.
Note: Prepare similar concentrations (µg/µl) of the different types of HS and CS disaccharides as stock solutions, and mix these to generate HS and CS standards. Add a multiplying factor to the D-7000 HSM software in order to account for the different molecular masses of the disaccharides. - In our hands, background noise in the HS samples (absent from the CS samples) requires the non-enzyme treated sample to be used as background control; subtract this from the chromatogram of the enzyme treated sample.
- Run treated and non-treated samples directly following each other to minimize changes in buffer conditions between the runs.
- The D-7000 HSM software exports data into an Excel file. We recommend to use a ‘macro’ (set of programming instructions for Excel) to calculate the total pmol per peak based on the signal for the relevant standard. A chromatogram as well as the resulting calculations are shown in Figure 9.
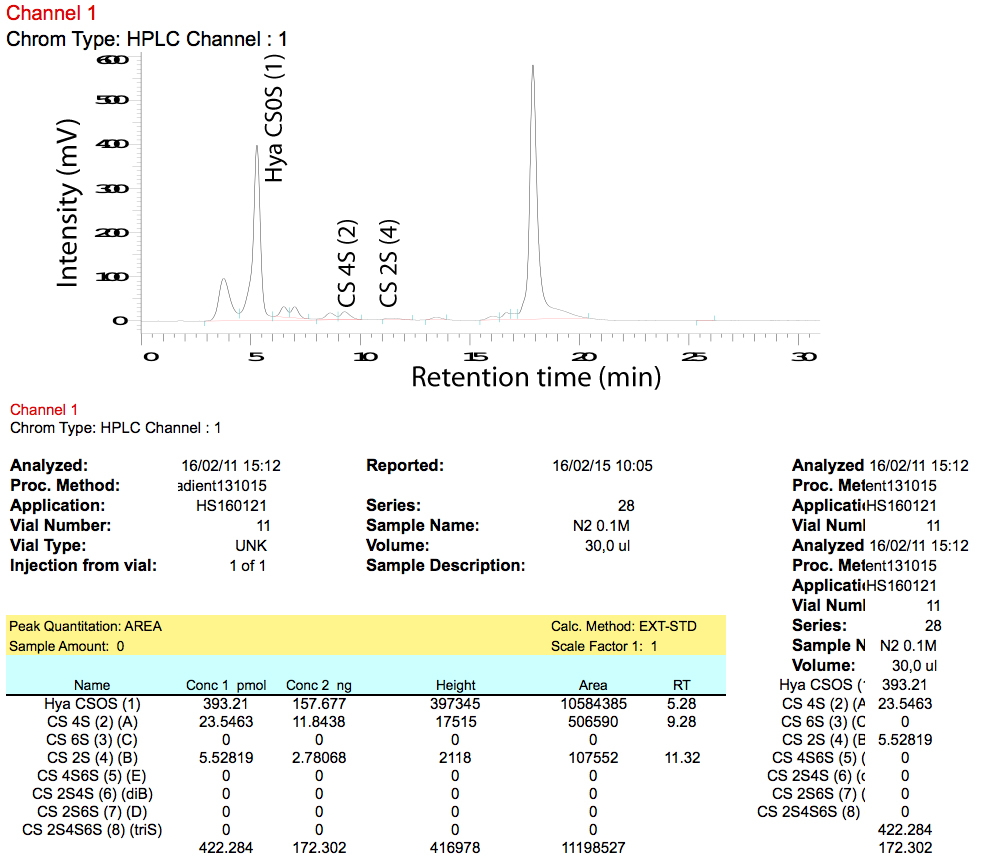
Figure 9. Chromatogram (upper panel) and calculations (lower panel) as exported by D-7000 HSM software - Use total pmol values to calculate the relative amount of a certain disaccharide (disaccharide composition) in each sample.
- The total sulfation is calculated based on the relative amount of sulfated disaccharides, and is therefore given as sulfation per 100 disaccharides.
- In order to quantify the total recovery from a given sample the amount (in pmol) of GAG recovered can be normalized to mg dry weight, after correction for the proportion of injected sample.
Note: In general, 10-90 µl were injected to ensure that the column was not overloaded. - As an alternative to dry weight, samples can also be normalized to DNA content before Benzonase treatment (see Procedure C, step C7). We use Quant-iTTM Broad Range Assay Kit (purchased from Molecular Probes) according to the manufacturer’s instructions and are satisfied with the results using this method.
- When comparing wild type and mutant strains we use samples that are purified and analyzed in parallel. We consider samples grown and isolated on different days as independent experiments and normally conduct at least three independent experiments.
- Use disaccharide standards analyzed in the same sample series to identify and quantify the peaks based on their elution position and peak size using the D-7000 HPLC System Manager (HSM) software version 4.1.
- Statistical analysis and data presentation
- All graphs are generated using GraphPad Prism version 5.0b for Mac OS X (GraphPad Software, San Diego California USA, www.graphpad.com).
- Diagrams show mean values and error bars define standard error of mean (SEM).
- One-tailed Mann-Whitney tests are performed where possible and P-values < 0.05 are considered significant.
- All graphs are generated using GraphPad Prism version 5.0b for Mac OS X (GraphPad Software, San Diego California USA, www.graphpad.com).
Notes
- Growth conditions will affect GAG composition, most likely due to changes in GAG structures during C. elegans development. We therefore suggest to only compare worms that have been grown under similar conditions. Comparing animals of the same developmental stage strongly improved reproducibility in our hands.
- Even though NAP-10 columns can be reused and should not retain high molecular weight molecules, we suggest reusing columns only for similar samples and not for material from other species to avoid contamination.
- When analyzing the chain length of GAGs from C. elegans we noted that they appeared to be shorter than those from mammals. The protocol presented here might therefore need optimization for separation of sulfated and non-sulfated GAGs from other species.
Recipes
Note: Unless noted otherwise buffers were prepared with non-autoclaved, deionized water and stored at room temperature. Sterilized buffers were either autoclaved (120 °C, 20 min) or passed through a 0.45 µm filter.
- Media for C. elegans growth and handling
- 1 M CaCl2
1.47 g CaCl2
Adjust volume to 10 ml - 5 N KOH
14.028 g KOH
Adjust volume to 50 ml - 1 M MgSO4
12.324 g MgSO4
Adjust volume to 50 ml - Cholesterol solution
5 mg/ml in 99.5% ethanol
Sterilize by filtration - 1 M potassium phosphate buffer pH 6.0 (KPO4)
120 g KH2PO4
27.55 g K2HPO4•3H2O per 1 L
Adjust pH to 6.0 with 1 M K2HPO4
Autoclave - Rich Nematode Growth medium (RNGM)
1.5 g NaCl
7.5 g agarose
3.75 g Bacto peptone per 500 ml
Autoclave
Cool to less than 55 °C and then add:
500 µl cholesterol
500 µl 1 M MgSO4
500 µl 1 M CaCl2
12.5 ml 1 M KPO4 buffer
Mix carefully before dispensing into Petri culture dishes of the desired size
Dry plates at room temperature and store at 4 °C - LB agar
2.5 g bacteriological agar
5 LB tablets per 250 ml
Autoclave
Store at 4 °C - 2x YT medium
1.6 g Bacto peptone
1 g Bacto yeast extract
0.5 g NaCl per 100 ml
Autoclave
Store at 4 °C - M9 buffer
5 g NaCl
15.12 g Na2HPO4•7H2O
3 g KH2PO4 per 1 L
Autoclave
Add 1 ml 1 M MgSO4
- 1 M CaCl2
- Buffers for GAG purification
Note: All buffers for washing and elution from DEAE columns were filtered through 0.45 µm filters.- 20% ethanol
100 ml 99% ethanol
Adjust volume to 500 ml - 1 M MgCl2
2.03 g MgCl2
Adjust volume to 10 ml - Stock solutions
- 10% Triton X-100
5 ml Triton X-100 (careful, very viscous!)
Adjust volume to 50 ml - 5 M NaCl
292.2 g NaCl
Adjust volume to 1 L
Note: Filtering the 5 M NaCl is an alternative to filtering all DEAE buffers. - 1 M CaCl2 (see Recipes A1)
- 1 M Tris-HCl
121.14 g Tris
Adjust pH to 7.5 with 6 N HCl
Adjust volume to 1 L
Autoclave
- 10% Triton X-100
- Protease buffer (50 mM Tris/HCl pH 8.0, 1 mM CaCl2, 1% Triton X-100)
5 ml 1 M Tris-HCl
100 µl 1 M CaCl2
10 ml 10% Triton X-100 on 80 ml ddH2O
Adjust pH with 6 M HCl
Adjust volume to 100 ml
Store at 4 °C - DEAE elution buffer (1.5 M NaCl)
- DEAE wash buffers (0.1 M NaCl or 0.25 M NaCl)
- DEAE wash buffer, pH 8 (50 M Tris/HCl pH 8.0, 0.1 M NaCl)
25 ml 1 M Tris-HCl
10 ml 5 M NaCl on 400 ml ddH2O
Adjust pH with 6 M HCl
Adjust volume to 500 ml - DEAE wash buffer, pH 4 (50 mM NaOAc pH 4.0, 0.1 M NaCl)
25 ml 1 M NaOAc
10 ml 5 M NaCl on 400 ml ddH2O
Adjust pH with 1 M acetic acid
Adjust volume to 500 ml - 0.1% AgNO3
100 mg AgNO3
Adjust volume to 100 ml - 5x chondroitinase ABC buffer (0.2 M Tris-acetate pH 8.0)
1.211 g Tris in 40 ml ddH2O
Adjust pH with 1 M acetic acid
Adjust volume to 50 ml - 2x heparinase lyase buffer (10 mM HEPES pH 7.0, 2 mM CaCl2)
119.2 mg HEPES
100 µl 1 M CaCl2 on 40 ml ddH2O
Adjust pH with 6 M HCl
Adjust volume to 50 ml
- 20% ethanol
Acknowledgments
Funding from the Swedish Research Council (to L.K.), the German Academic Exchange Service (to T.D.) and Stiftelsen för Proteoglykanforskning (to L.K.) are acknowledged. GAG purification was described by Ledin et al., JBC 2004 (doi: 10.1074/jbc.M405382200) and the protocol modified for C. elegans was first published by Dierker et al., Sci. Rep. 2016 (doi: 10.1038/srep34662).
References
- Bishop, J. R., Schuksz, M., and Esko, J. D. (2007). Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446(7139): 1030-1037.
- Bulow, H. E., and Hobert, O. (2006). The molecular diversity of glycosaminoglycans shapes animal development. Annu Rev Cell Dev Biol 22: 375-407.
- Dagalv, A., Holmborn, K., Kjellen, L., and Abrink, M. (2011). Lowered expression of heparan sulfate/heparin biosynthesis enzyme N-deacetylase/n-sulfotransferase 1 results in increased sulfation of mast cell heparin. J Biol Chem 286(52): 44433-44440.
- Dierker, T., Shao, C., Haitina, T., Zaia, J., Hinas, A., and Kjellen, L. (2016). Nematodes join the family of chondroitin sulfate-synthesizing organisms: Identification of an active chondroitin sulfotransferase in Caenorhabditis elegans. Sci Rep 6: 34662.
- Holmborn, K., Habicher, J., Kasza, Z., Eriksson, A. S., Filipek-Gorniok, B., Gopal, S., Couchman, J. R., Ahlberg, P. E., Wiweger, M., Spillmann, D., Kreuger, J., and Ledin, J. (2012). On the roles and regulation of chondroitin sulfate and heparan sulfate in zebrafish pharyngeal cartilage morphogenesis. J Biol Chem 287: 33905-33916.
- Kiselova, N., Dierker, T., Spillmann, D., and Ramstrom, M. (2014). An automated mass spectrometry-based screening method for analysis of sulfated glycosaminoglycans. Biochem Biophys Res Commun 450: 598-603.
- Lawrence, R., Olson, S. K., Steele, R. E., Wang, L., Warrior, R., Cummings, R. D., and Esko, J. D. (2008). Evolutionary differences in glycosaminoglycan fine structure detected by quantitative glycan reductive isotope labeling. J Biol Chem 283(48): 33674-33684.
- Ledin, J., Staatz, W., Li, J. P., Götte, M., Selleck, S., Kjellén, L., and Spillmann, D. (2004). Heparan sulfate structure in mice with genetically modified heparan sulfate production. J Biol Chem 279(41): 42732-42741.
- Shao, C., Shi, X., Phillips, J. J., and Zaia, J. (2013). Mass spectral profiling of glycasaminoglycans from histological tissue surfaces. Anal Chem 85(22): 10984-10991.
- Townley, R. A. and Bulow, H. E. (2011). Genetic analysis of the heparan modification network in Caenorhabditis elegans. J Biol Chem 286:16824-16831.
- Toyoda, H., Kinoshita-Toyoda, A., and Selleck, S. B. (2000). Structural analysis of glycosaminoglycans in Drosophila and Caenorhabditis elegans and demonstration that tout-velu, a Drosophila gene related to EXT tumor suppressors, affects heparan sulfate in vivo. J Bioll Chem 275(4): 2269-2275.
- Yamada, S., Sugahara, K., and Ozbek, S. (2011). Evolution of glycosaminoglycans. Commun Integr Biol 4(2): 150-158.
- Yamada, S., Van Die, I., Van den Eijnden, D. H., Yokota, A., Kitagawa, H., and Sugahara, K. (1999). Demonstration of glycosaminoglycans in Caenorhabditis elegans. FEBS Lett 459(3): 327-331.
- Zhang, L. (2010). Progress in molecular biology and translational science. Elsevier 93: 1-17.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Dierker, T. and Kjellén, L. (2017). Separation and Purification of Glycosaminoglycans (GAGs) from Caenorhabditis elegans. Bio-protocol 7(15): e2437. DOI: 10.21769/BioProtoc.2437.
Category
Biochemistry > Carbohydrate > Polysaccharide
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.