- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

RNA Interference Screening to Identify Proliferation Determinants in Breast Cancer Cells
Published: Vol 7, Iss 15, Aug 5, 2017 DOI: 10.21769/BioProtoc.2435 Views: 9624
Reviewed by: Aswad KhadilkarVarpu MarjomakiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2016

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
RNAi screening technology has revealed unknown determinants of various biological signaling pathways in biomedical studies. This protocol provided detailed information about how to use RNAi screening to identify proliferation determinants in breast tumor cells. siRNA-based libraries targeting against Estrogen receptor (ER)-network, including 631 genes relevant to estrogen signaling, was constructed for screening in breast cancer cells. Briefly, reverse transfection of siRNA induced transient gene knockdown in MCF7 cells. First, the transfection reagent for MCF7 cells was selected. Next, the Z’-score assay was used to monitor if screening conditions yielded efficiently. Then, the ER-network siRNA library screening was preceded by automatic machines under optimized experimental conditions.
Keywords: RNA interference (RNAi)Background
RNA interference (RNAi) is a biological process that can be exploited to inhibit gene expression by causing the destruction of specific mRNA molecules. Knockdown of specific genes by RNAi technology is often associated with phenotypic changes, which has made RNAi widely used in life science research. Two systems are utilized for high-throughput RNAi screening, one is lentivus-based short hairpin RNA (shRNA) library screening; the other is chemical synthesized small interference RNA (siRNA)-based screening (Boutros et al., 2008). shRNA-based transfection induces stable gene knockdown in cells. siRNA-based transfection induces transient gene knockdown. Lentiviral pooled shRNA libraries contain lentiviruses with shRNAs targeting against either genomic DNA or a group of genes. Following analysis is required to distinguish target genes after screening, such as chip-based DNA microarray or next generation sequencing (NGS). However, in siRNA-based libraries, siRNAs against each single target gene are distributed in each well of 96-well or 384-well plates. A siRNA library may include many plates depending on the number of targeting genes in this library. For siRNA library screening, no further techniques are required to identify targeting genes.
In our studies, we designed the Estrogen receptor (ER)-network around 5 seed proteins relevant to estrogen signaling: the ER genes ESR1 (ERα) and ESR2 (ERβ), the estrogen-related receptors ESRRA and ESRRG, and CYP19A1 (aromatase). 631 genes were selected as ER network. Next, we constructed siRNA-based libraries targeting against ER network genes into 96-well plates, which were custom-made from QIAGEN (MD, USA). siRNAs against those genes were distributed into 11 x 96-well plates. Two siRNAs were selected for each gene and mixed in one well (Zhang et al., 2016). The advantage of our method provides high-throughput screening by using automatic machines (Cybio, Combi-nL or Wellmate dispenser) to dispense liquid to speed the screening process.
Different types of cancer cell lines had been used in RNAi screening with our methods (Astsaturov et al., 2010; Murray et al., 2014; Zhang et al., 2016), such as estrogen positive breast cancer MCF7, estrogen-independent MCF7 (LCC1 and LCC9), triple negative breast cancer MDA-MB-231, epidermoid cancer A431 and human fibroblast HFF1 cells etc. For each cell line, the optimal transfection reagent has to be determined before RNAi library screening. Z’-score is taken as a quantitative parameter to control the experiment quality for various cell lines and corresponding transfection reagents. In this assay, we utilize ER-network RNAi screening in MCF7 cells as an example to describe the protocol (Zhang et al., 2016). It also fits other cell lines or other gene network RNAi library with minor modification, such as type of transfection reagent, cell plating density, Cell Titer blue incubation time or RNAi library scale (total number of siRNA library plates), which will be noted. In this article, these protocols will be described in three parts: 1) Selection of transfection reagents; 2) Z’-score determination; 3) Screening an RNAi library.
Materials and Reagents
- Pipette tips for CyBi-Well Vario 96 channel simultaneous Pipettor (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 5587 )
- V-bottom 96-well plates (Corning, catalog number: 3357 )
- Flat-bottom 96-well plates (Corning, catalog number: 3595 )
- 50 ml conical tube
- Corning 0.22 µm vacuum filter system (Corning, catalog number: 431098 )
- T75 flasks (Corning, Costar)
- Labels with Barcode
- MCF7 cells (Tissue Culture Shared Resource, Lombardi Cancer Center, Georgetown Univ.)
- AllStars Negative Control siRNA (QIAGEN, catalog number: 1027281 )
- AllStars Hs Cell Death siRNA (QIAGEN, catalog number: 1027299 )
- AP2A siRNA (QIAGEN, catalog number: SI04371283 )
- GRB14 siRNA (QIAGEN, catalog number: SI00430703 )
- Opti-MEM reduced serum medium (Thermo Fisher Scientific, GibcoTM, catalog number: 31985070 )
- IMEM medium (Mediatech, catalog number: 10-024-CV )
- Trypsin-EDTA (0.5%), no phenol red (Thermo Fisher Scientific, GibcoTM, catalog number: 15400054 )
- Charcoal-stripped bovine calf serum (CCS) (Gemini Bio-Products, catalog number: 100-213 )
- Estradiol (Sigma-Aldrich, catalog number: E8875 )
- Cell Titer Blue (Promega, catalog number: G8082 )
- Hank’s balanced salt solution (HBSS) without calcium, magnesium, phenol red (GE Healthcare, HycloneTM, catalog number: SH30588.01 )
- ER network siRNA library plates (Customized from QIAGEN)
- siRNA suspension buffer (QIAGEN)
- Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific, InvitrogenTM, catalog number: 13778500 )
- HiPerfect (QIAGEN, catalog number: 301704 )
- Dharmafect 1-4 transfection reagent (GE Dharmacon, catalog numbers: T-2001 , T-2002 , T-2003 , T-2004 )
- RNAiFect (QIAGEN)
- 70% (v/v) ethanol (filtered via Corning 0.22 µm vacuum filter system)
- 0.22 µm filtered ddH2O
Equipment
- CyBi-Well Vario 96 channel simultaneous Pipettor (CyBio)
- Multidrop Combi-nL reagent dispenser (Thermo Fisher Scientific, catalog number: 5840400 )
- WellMate microplate dispenser (Thermo Scientific Matrix)
- AccuSpin 3R Centrifuge with Ch.003741 rotor (Thermo Fisher Scientific, catalog number: 4393 ) and swing rectangular buckets with adapters (Thermo Fisher Scientific, catalog number: 75006449 )
- Magnetic stirrer (Thermo Fisher Scientific)
- Envision multi-label plate reader with 560Ex/590Em filter set (PerkinElmer, catalog number: 2104-0010 )
- 500 ml glass bottle (Corning, Costar)
Part I. Selection of transfection reagents
Procedure
Transfection with multiple lipids (transfection reagents) in MCF7 cells
- Transfect cells in 96-well plate in 7 blocks, for each block (12 wells):
3 wells: lipid + Opti-MEM
3 wells: 20 nM AllStars negative control siRNA (QIAGEN, MD) + lipid + Opti-MEM
3 wells: 20 nM AllStars Death control siRNA (QIAGEN, MD) + lipid + Opti-MEM
3 wells: 20 nM AP2A1 siRNA (QIAGEN, MD) + lipid + Opti-MEM - For lipid: diluted lipid (recipe as in Table 1) will be added (15 µl/well) after appropriate dilution in Opti-MEM (Invitrogen, MD). Diluted lipid will be aliquot into 12 wells.
Table 1. Recipe of diluted lipid. To account for pipetting loss, 187.5 µl (12.5 x 15 µl) diluted lipid will be made in Opti-MEM.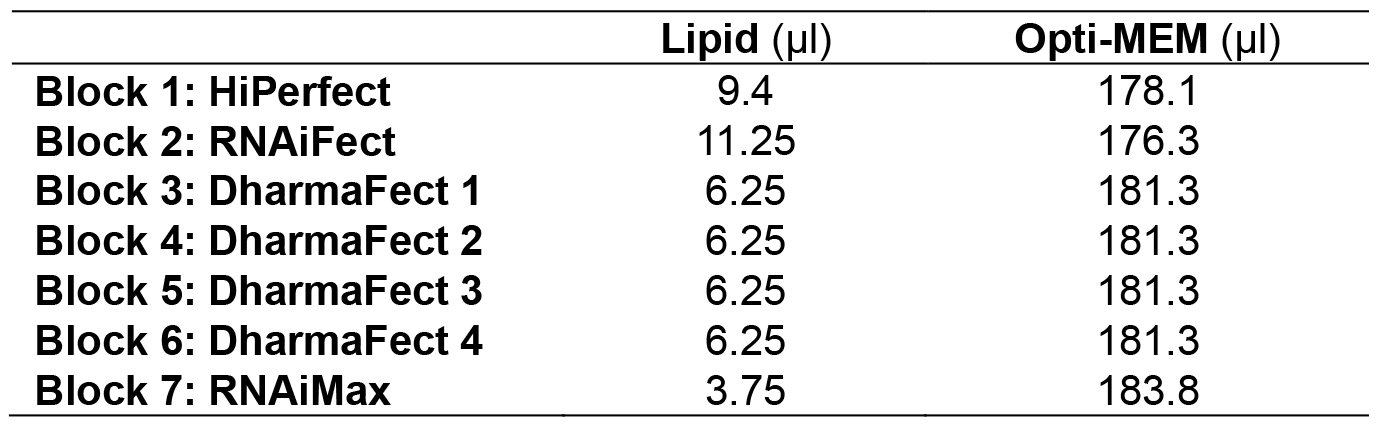
- For siRNA: 1 µM siRNA is diluted in Opti-MEM (1:3) and 7 µl diluted siRNA is added to each corresponding well. Each siRNA is dispensed into 21 wells, therefore, 147 µl are needed. To account for pipetting loss, we made 165 µl diluted siRNA: 55 µl 1 µM siRNA + 110 µl Opti-MEM.
- Split cells, count and calculate dilution for 8,000 cells per well in 100 µl IMEM + 5% charcoal-stripped bovine calf serum (CCS) + 1 nM estradiol (Sigma-Aldrich, MI).
- Set up a 96-well plate:
- Pipette siRNA (Opti-MEM for lipid control wells: A1-12, E1-9), 7 µl/well to following wells: siNEG, wells B1-12 and F1-9; siAP2A1, wells C1-12 and G1-9; siDEATH, wells D1-12 and H1-9.
- Pipette lipid mixture (Opti-MEM for medium control wells: E10-12, F10-12, G10-12 and H10-12), 15 µl/well to following wells: A1-12, B1-12, C1-12, D1-12, E1-9, F1-9, G1-9 and H1-9 (see Figure 1 for plate layout).
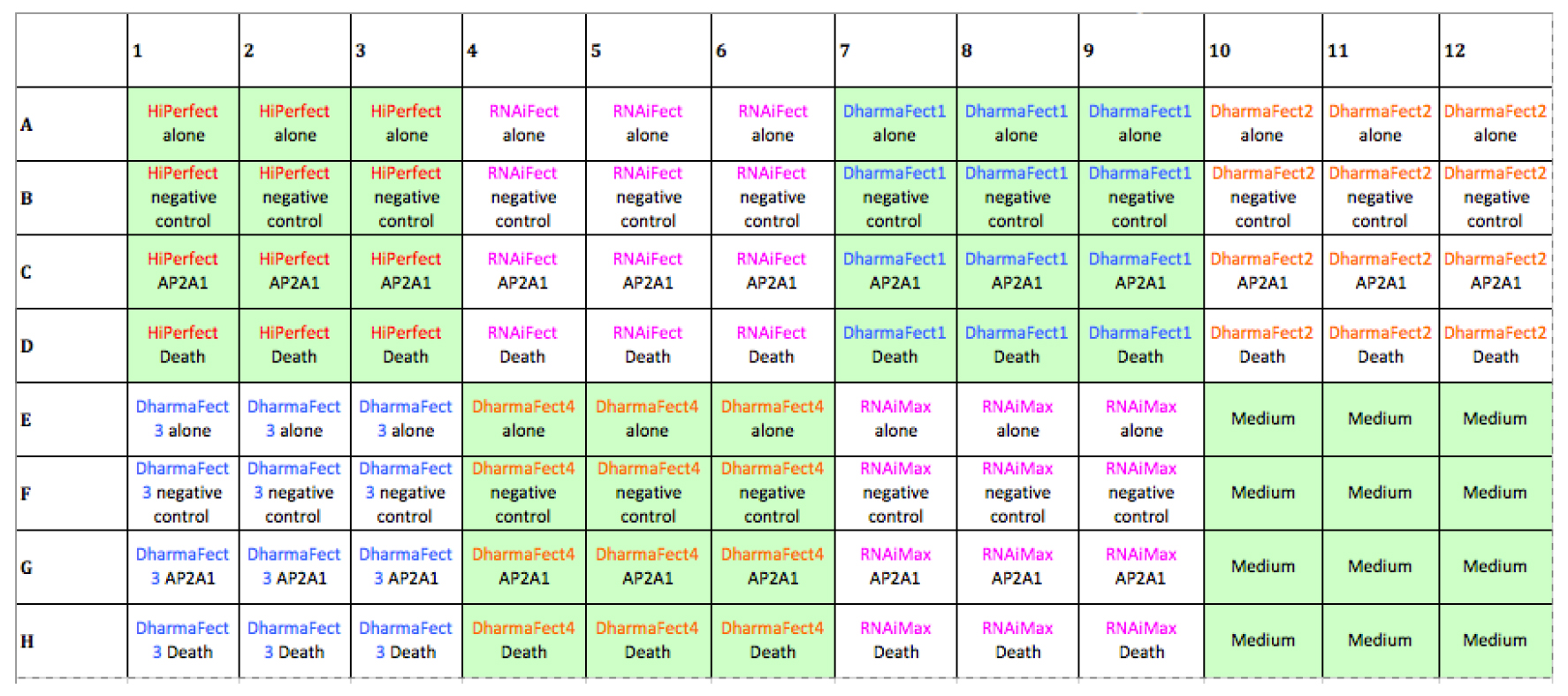
Figure 1. Layout of transfection reagent selection plate. Efficiency of a variety of transfection reagents was tested in MCF7 cells. - Incubate for 20 min at room temperature, next add 100 µl MCF7 cells/well with WellMate Microplate dispenser (Thermo Scientific Matrix, USA), then incubate at 37 °C, 5% CO2.
- Five days later, add 20 µl of 1:1 mixture of Cell Titer Blue:HBSS to each well and incubate at 37 °C to allow cells to convert resazurin to resorufin. The fluorescent signal is measured by Envision multi-label plate reader (with excitation wavelength 560 nm/emission wavelength 590 nm) every hour up to 4 h. For this experiment, 2 h is typically the optimal time point to read out with 4 h nearing the maximum signal of the assay where dynamic range is not compromised.
Data analysis
Normalize all cell growth to control cells (in wells E, F, G and H10-11). Next, assess the growth inhibition induced by negative control siRNAs (siNEG, in wells B and F), death control siRNAs (siDEATH, in wells D and H), and AP2A1 siRNA (in wells C and G). Then, compare the effect of various transfection reagents. Viability of AP2A1 siRNA should be within the middle of the dynamic range between siNEG and siDEATH. Select the lipid mixture containing the transfection reagent that provides not only the highest viability with siNEG, but also the lowest viability with siDEATH.
Part II. Z’-score determination
Procedure
Day 1
Set up a siRNA Z’-score plate in V-bottom 96-well plate containing 0.24 µM siNEG and siDEATH (using layout shown in Figure 2). To make 0.24 µM siRNA solution: 72 µl 20 µM siRNA is mixed in 5,928 µl siRNA suspension buffer; or 1,440 µl 1 μM siRNA is mixed in 4,560 µl siRNA suspension buffer. One hundred µl of diluted siNEG or siDEATH (0.24 µM) is dispensed into each well (using layout shown in Figure 2). Then siRNA Z’-score plate is frozen at -20 °C for later use. 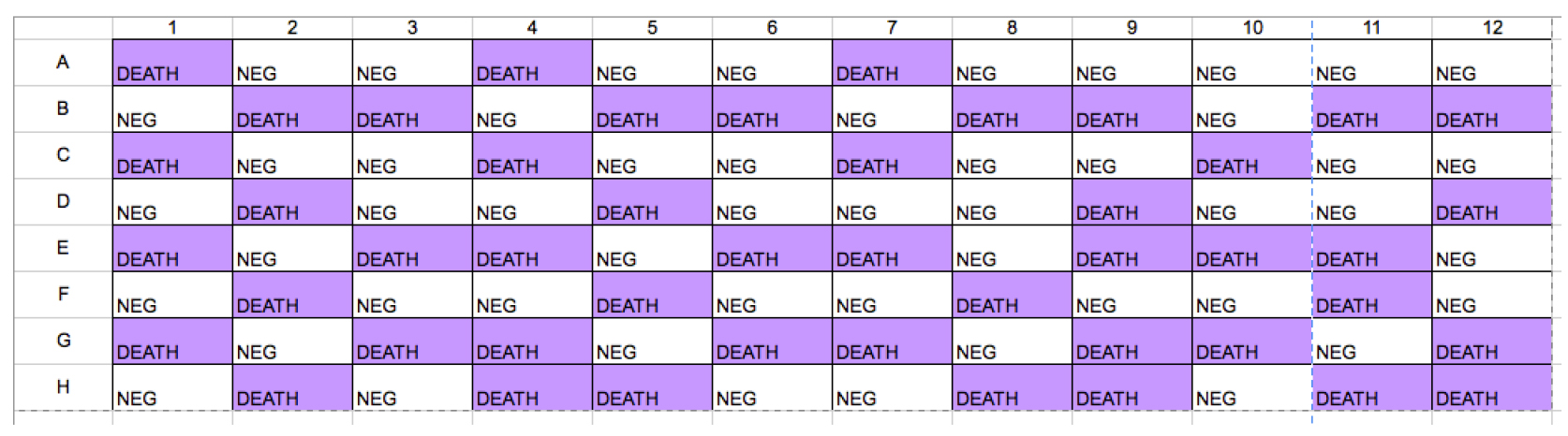
Figure 2. Layout of siRNA Z’-score plate
Day 2
Each experimental plate (96-well plate, Costar, Corning, USA) must have 10.5 µl of diluted lipid transfection reagent, 10 µl of siRNA and 8,000 cells per well. Two replicate plates are run at one experiment. For two plates (192 wells), 2,016 µl transfection reagents are required. Due to the loss of machine priming, 3 ml of total transfection reagent is loaded on Combi-nL machine that aliquot 10.5 µl to each well in two experimental plates. After thawing at room temperature, pipette 10 µl from the siRNA Z’-score plate into each experimental replicate plate containing 10.5 µl of diluted transfection reagent in each well on CyBio machine. The final concentration of siRNA in cells is 20 nM. Next, cells will be added into experimental plates for culture by Wellmate microplate dispenser.
Procedure and timeline
- Clean WellMate microplate dispenser with 15 ml of 70% ethanol (pre-filtered by 0.22 µm vacuum filter), then 15 ml of ddH2O (pre-filtered by 0.22 µm vacuum filter), and lastly 15 ml of IMEM (no serum).
- Split cells and count. Dilute 8,000 cells in 100 µl for one well (For one 96-well plate, 80,000 cells/ml, need 16 ml, prepare 40 ml in a 50 ml conical tube). Plate cells in new flasks if necessary.
- Dispense 15 ml Opti-MEM in a 50 ml conical tube.
- Clean Combi-nL with 7 ml of filtered (0.22 µm) 70% ethanol, 7 ml distilled water (0.22 µm filtered), 7 ml Opti-MEM.
- Take siRNA Z’-score plate out from -20 °C and thaw plate at room temperature. Next, spin plate in centrifuge to get liquid in center of wells at 3,500 rpm (1,935 x g), 5 min, room temperature, take plate out immediately to keep condensation from forming.
- Dilute transfection reagent: 144 µl in 2.86 ml Opti-MEM in a 50 ml conical tube.
- Dispense 10.5 µl/well of diluted transfection reagent to the each Costar 96-well plate (experimental plate) by Combi-nL machine.
- Start Cybio machine, load plates and run program (Figure 3) to distribute siRNAs from siRNA Z’-score plate to experimental plate (pre-loaded with lipid from step 2g). The loading position for plates:
- siRNA Z’-score plate–loaded on stack A (left arm) on CyBio.
- Experimental plate–loaded on stack A (right arm, with diluted lipid) on CyBio.
- siRNA Z’-score plate–loaded on stack A (left arm) on CyBio.
- Wait for 10 min at room temperature for siRNA-lipid complexes to form in experimental plate. While waiting, set up dispensing program on the WellMate dispenser.
- Use the WellMate microplate dispenser to dispense 100 µl cells/well into each experimental plate, incubate experimental plate at 37 °C, 5% CO2.
- Clean WellMate dispenser with 15 ml filtered ddH2O, then 15 ml filtered 70% ethanol, switch off machine.
- Clean the Combi-nL dispenser with 7 ml distilled water, and 7 ml of filtered (0.45 µm) 70% ethanol, switch off machine.
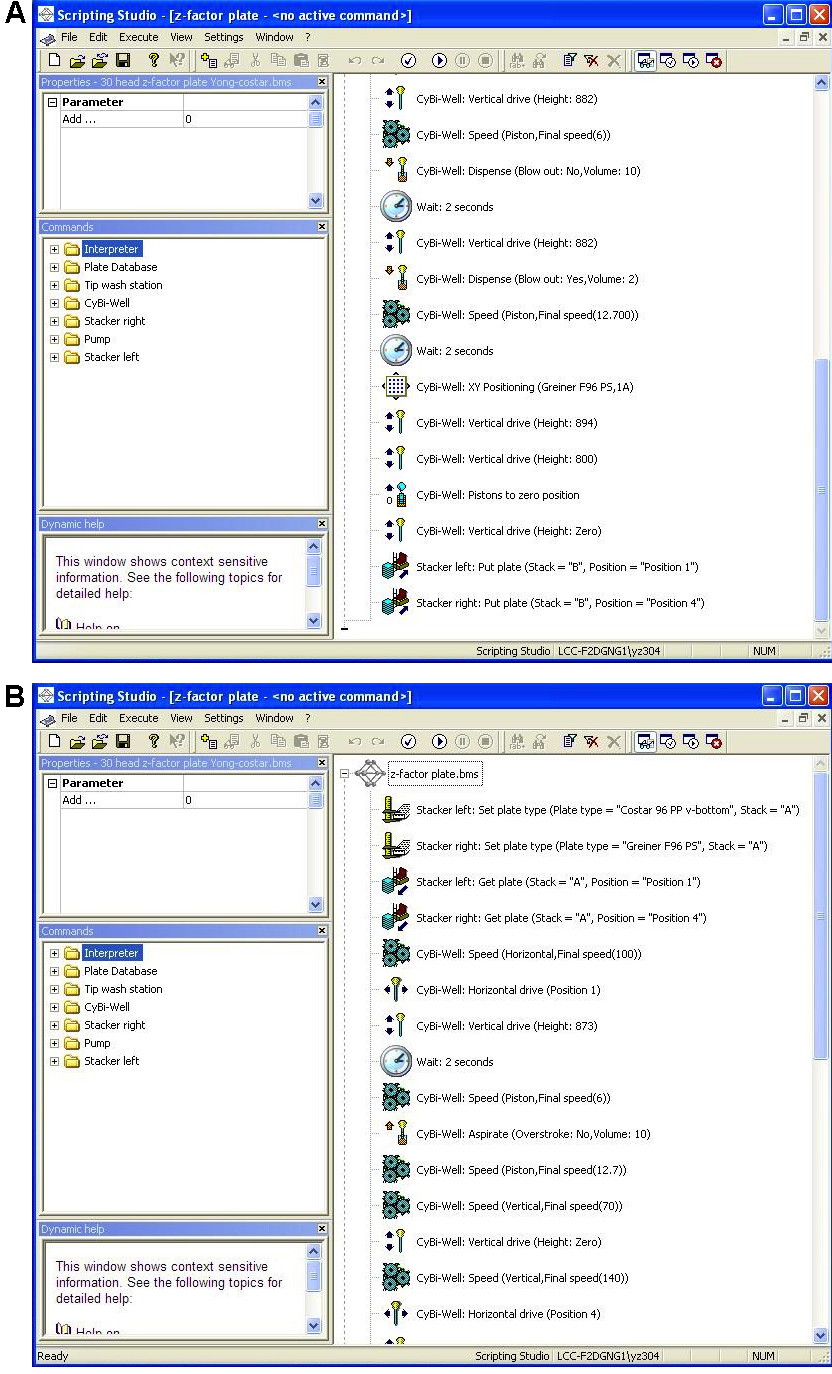
Figure 3. Cybio program to dispense siRNA into experimental plates. A. The first part of program; B. The second part of program.Day 7
Add 20 µl of 1:1 mixture of Cell Titer Blue:HBSS to each well and read out every hour up to 4 h (as described above).
Data analysis
- Based on Cell-Titer Blue read out, calculate the average viability value of siNEG and siDEATH. Calculate Z’-score: Z’-score = 1 - (3 x S.D. of siDEATH + 3 x S.D. of siNEG)/(Average viability reading of siNEG - Average viability reading of siDEATH) (Zhang et al., 1999; Birmingham et al., 2009). The reasonable range of Z’-score is between 0.7-1. If Z’-score is smaller than 0.7, re-assess assay parameters and repeat the Z’-score experiment. Once Z’-score is within range, continue with large scale RNAi library screening.
- A representative viability measurement (Figure 4A) and dot plots (Figure 4B) from one Z’-score test were shown here. In this experiment, average viability reading of siNEG, siDEATH and corresponding standard deviation values (S.D.) were obtained from Figure 4A. Then Z’-score was calculated as 0.81, which was in the range of 0.7-1, therefore, assay parameters used in this experiment were optimal for siRNA library screening.
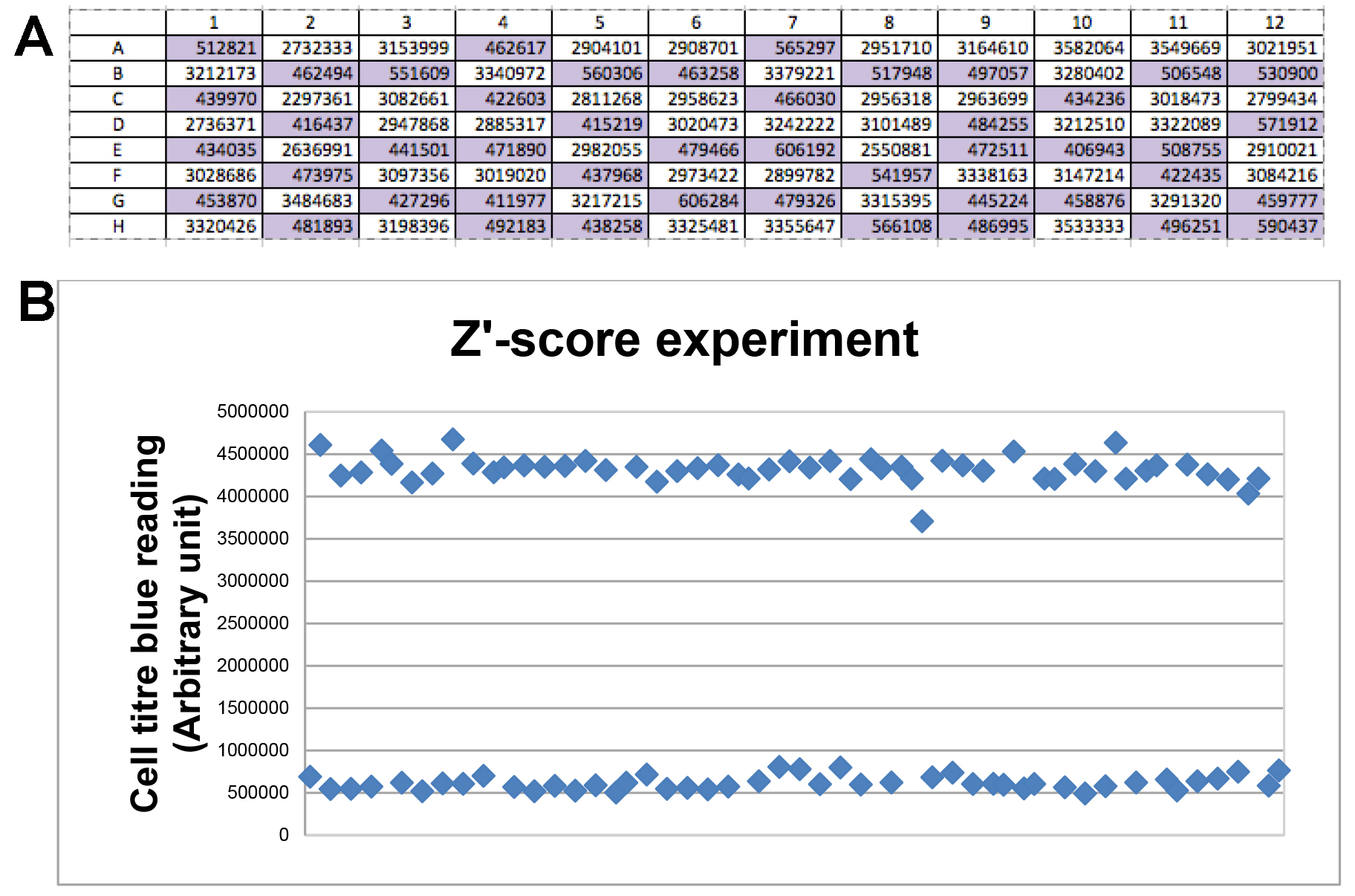
Figure 4. Representative data of Z’-score test. A. Viability measurement by Cell Titre Blue reading. Purple wells: siDEATH-transfected cells; white wells: siNEG-transfected cells. B. Dot plots of viability measurements. Each dot indicated the value of cell tire blue reading in each well. Upper line of dots indicated those wells containing siNEG-transfected cells; lower line of dots indicated those wells with siDEATH-transfected cells.
Part III. Screening an RNAi library
Procedure
- SiRNA preparation for RNAi screening
- In this article, we utilize an ER-network RNAi library screening as an example to describe the protocol. The ER network siRNA library contains siRNAs against 631 genes, which were custom-made from QIAGEN (MD, USA). siRNAs against those genes were distributed into 11 x 96-well plates. Two siRNAs were selected for each gene and mixed in one well.
Notes: For other siRNA library screening, the total number of plates will be determined based on the number of targeted genes and number of targeting siRNAs per gene as well. In summary, the total number and final layout of wells containing siRNA in an RNAi library will modify the screening protocol. - Calculate the amount of various control siRNA (0.24 µM) needed to make 11 plates (100 µl each well):
- Preparation of negative control siRNA (siNEG)
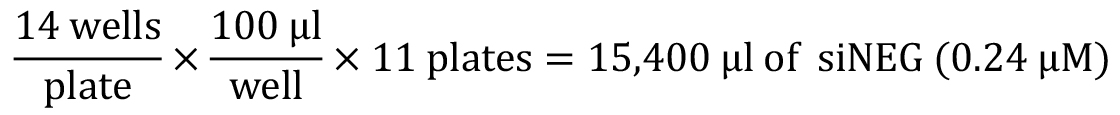
Accounting for pipetting error, 18 ml of 0.24 µM siNEG will be prepared as followed: 4.32 ml siNEG (1 µM) + 13.68 ml siRNA suspension buffer. - Preparation of DEATH, AP2A1(X) and GRB14(Y) siRNA
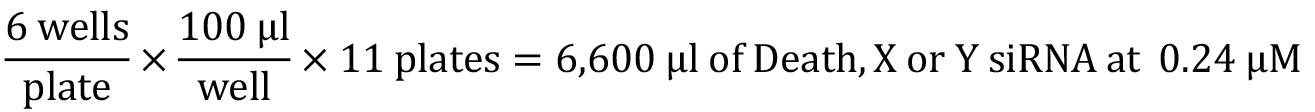
Accounting for pipetting error, 8 ml of 0.24 µM DEATH, X and Y siRNA will be prepared as followed: 1.92 ml DEATH, X or Y siRNA (1 µM) + 6.08 ml siRNA suspension buffer. - Preparation of ER network library siRNAs
For each siRNA mix in one well, prepare 100 µl 0.24 µM siRNA as followed: 24 µl 1 µM stock siRNA + 76 µl siRNA suspension buffer.
- In this article, we utilize an ER-network RNAi library screening as an example to describe the protocol. The ER network siRNA library contains siRNAs against 631 genes, which were custom-made from QIAGEN (MD, USA). siRNAs against those genes were distributed into 11 x 96-well plates. Two siRNAs were selected for each gene and mixed in one well.
- Estrogen Receptor siRNA Library Screening using RNAiMAX Transfection Reagent in MCF7 cells
Day 1
Autoclave 500 ml glass bottles and magnetic stir bars.
Day 2 - Set up siRNA Library plates (stock concentration: 0.24 µM, in V-bottom 96-well plates). Layouts are shown in the following 96-well plates (Plates #1-10, Figure 5; Plates #11, Figure 6). The total number of siRNA library plates is 11. Unlabeled wells are for siRNAs against ER network target genes. Labeled wells are for various controls (NEG, DEATH, AP2A1 and GRB14 and MOCK [siRNA suspension buffer]). 100 µl diluted siRNA was dispensed into each well, stock in -20 °C.
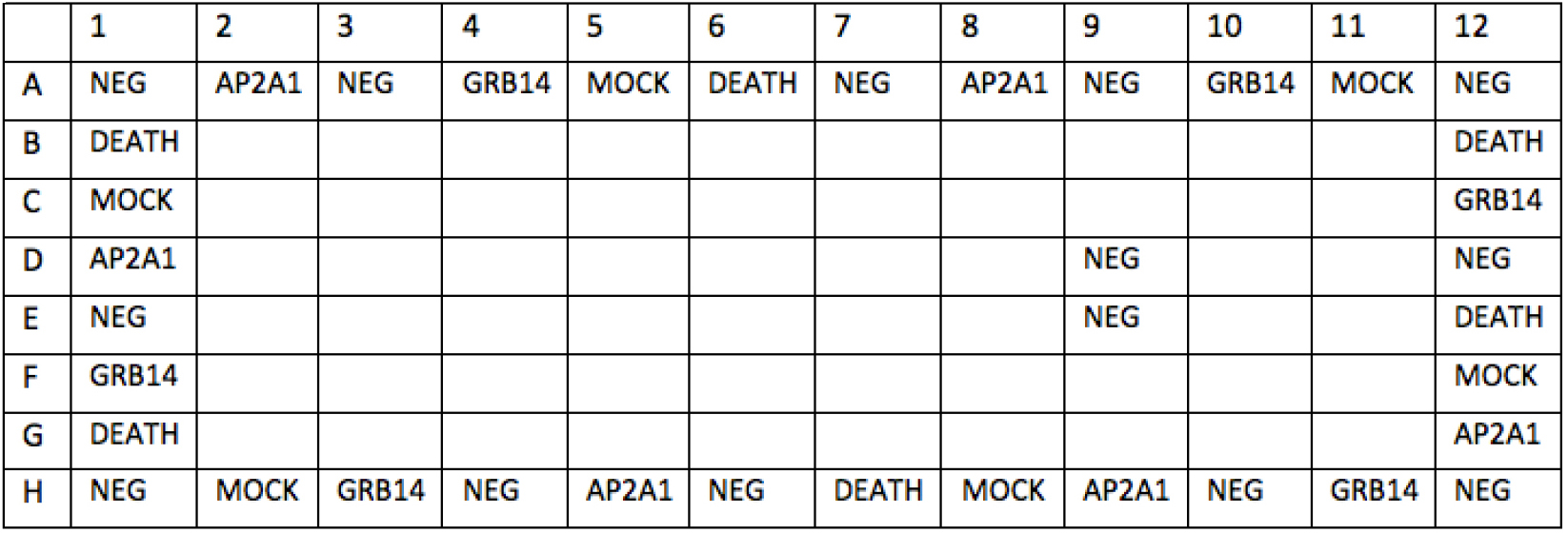
Figure 5. Layout of ER network siRNA library plate #1-10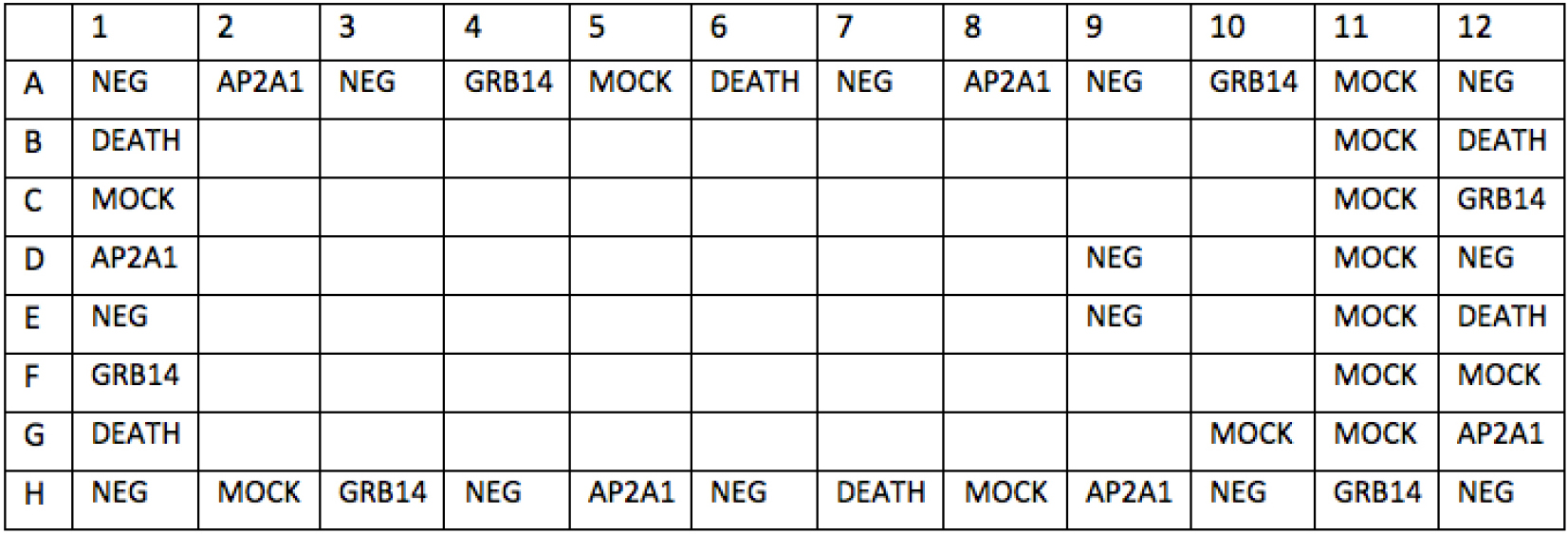
Figure 6. Layout of ER network siRNA library plate #11 - Next, prepare 11 flat-bottom 96-well plates as experimental plates. Barcodes for these 11 plates need to be printed and pasted on plates (front and middle). After labeling, these plates are stored in a cell culture hood overnight. Meanwhile, confirm that all materials, including screening media, transfection reagent, plates, transfer pipette tips, and cell cultures are ready.
- The Cybio program (Figure 7) will pipette 10 µl from 0.24 µM siRNA ER library plate in V-bottom plate, then mixed into Costar 96-well plate that already has 10.5 µl of diluted RNAimax transfection reagent. The final concentration of siRNA in cells was 20 nM.
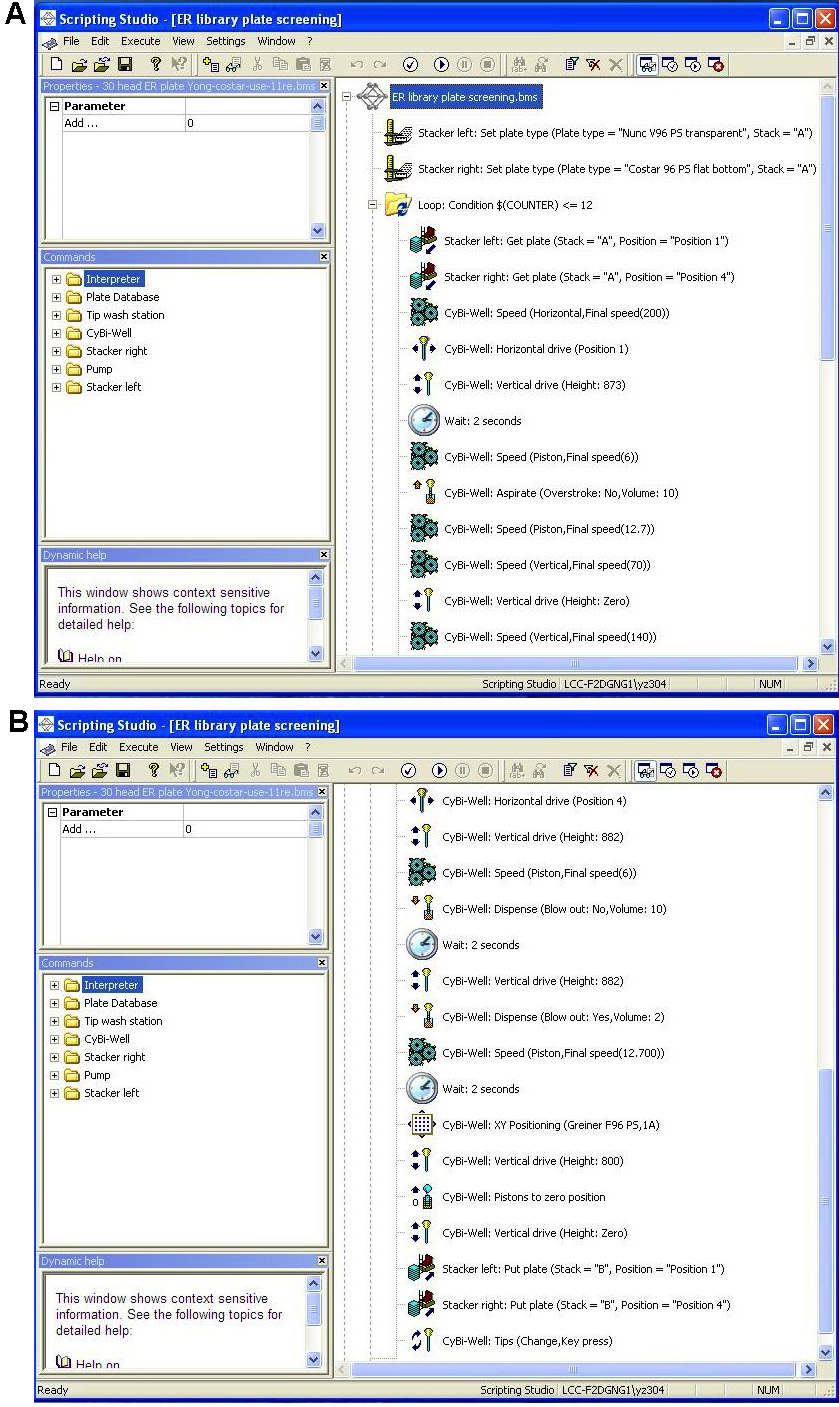
Figure 7. Cybio program to dispense siRNA into RNAi screening experimental plates. A. The first part of program; B. The second part of program. - Each experimental plate (Costar 96-well plate) must have 10.5 µl of diluted lipid transfection reagent (0.5 µl RNAimax + 10 µl Opti-MEM medium) added to each well with Combi-nL dispensing machine. 96 wells x 10.5 µl = 1,008 µl, 11 plates 11,088 µl. Accounting for loss from machine priming, make 15 ml for 11 plates. Then load these siRNA and experimental plates on Cybio machine as following:
- siRNA ER library plates (0.24 µM, 11 plates)–loaded on Left Arm, Stack A, plates ascending from the bottom with lids removed before loading;
- Experimental plates (11 plates)–loaded on Right Arm, Stack A; plates with ascending from the bottom with lids removed before loading.
Procedure and timeline:- Thaw siRNA library plates (stock concentration: 0.24 µM) at room temperature.
- Clean WellMate microplate dispenser machine with 15 ml of filtered 70% ethanol, then 15 ml of filtered ddH2O, and 15 ml IMEM medium (without serum).
- Split MCF7 cells and count. Dilute 8,000 cells in 100 µl (80,000 cells/ml). For one plate: 16 ml of diluted cells are needed. Therefore, for 11 plates: 250 ml diluted cells in 500 ml sterile bottle, with magnet stir bar on the magnetic stirrers, low speed. Plate cells in new flasks if necessary.
- Clean Combi-nL dispensing machine with 7 ml of filtered 70% ethanol, 7 ml filtered ddH2O, and 7 ml Opti-MEM medium.
- When the siRNA library plates have thawed, centrifuge them at 3,500 rpm (1,935 x g) for 5 min at room temperature. Take plate out immediately to keep condensation from forming.
- Dispense 15 ml Opti-MEM in a 50 ml conical tube.
- Dilute transfection reagent: 720 µl RNAimax in 14.3 ml Opti-MEM in a 50 ml conical tube.
- Dispense 10.5 µl/well of diluted transfection reagent to 11 experimental plates (Costar 96-well plates).
- Load experimental plates and siRNA plates on designated stacks of Cybio-machine. siRNA plates: Left Arm, Stack A; experimental plates: Right Arm, Stack A. Lids were removed before loading, and plates ascending from the bottom.
- Start Cybio machine, run program (Figure 7) to distribute 11 plates of library siRNAs as shown in attached plate layout (Figures 5 and 6). Set up a timer, record the time of each plate being processed. Proceed to the next step while completing this step.
Note: In this example, the time for running one plate and changing tips was 1 min and 15 sec. The total machine running process will take about 15 min. - After the first four experimental plates containing diluted siRNA-lipid mixture are completed, transfer these plates to the cell culture hood. A second person should continue running the remainder of plates on Cybio machine.
- After waiting 10-15 min from the time of siRNA-lipid mixture, use the WellMate microplate dispenser machine to dispense 100 µl of cells/well to the first four experimental plates. Wait for another 5 min, then prime the WellMate machine with 5-7 ml cell suspension, dispense cells for the next 4 plates; then wait for another 5 min, prime machine with 5-7 ml cell suspension, dispense cells for the last 3 plates. Document the time each plate received cells.
Notes:- Four plates as a group to be added with cells.
- Put WellMate dispenser probe into diluted cells only before adding cells into experimental plates, not earlier, and then prime machine with 5-7 ml cell suspension for each group.
- Four plates as a group to be added with cells.
- Clean WellMate dispenser with 15 ml filtered ddH2O, then 15 ml filtered 70% ethanol, and switch off the machine.
- Clean the Combi-nL dispenser with 7 ml distilled water, and 7 ml of filtered (0.45 µm) 70% ethanol, switch off machine.
Day 7
Add 20 µl of 1:1 mixture of Cell Titer Blue:HBSS to each well and read out every hour up to 4 h (as described above).
- siRNA ER library plates (0.24 µM, 11 plates)–loaded on Left Arm, Stack A, plates ascending from the bottom with lids removed before loading;
Data analysis
- Calculate median viability values (Cell Titer blue readout, arbitrary unit) of each control from the data of corresponding wells, mock (Plate 1-10: A5, A11, C1, F12, H2 and H8; Plate 11, A5, A-G11, C1, F12, H2, H8 and G10); siNEG (all plates: A1, A3, A7, A9, A12, E1, E9, D9, D12, H1, H4, H6, H10 and H12); siDEATH (all plates: B1, B12, A6, E12, G1 and H7); siAP2A1 (all plates: A2, A8, D1, G12, H5 and H9); siGRB14 (all plates: A4, A10, C12, F1, H3 and H11). Determine if AP2A1 or GRB14 siRNA yields median killing of cells, and if death control siRNA kills more than 90% of cells. If so, continue with following data processing and analysis. If not, go back to troubleshoot with technique.
- Calculate the viability index (VI) of each siRNA-transfected cell normalized to the median value of negative control siRNA-transfected cells:
VI = (viability of siRNA-transfected cell)/median viability of negative control-transfected cells - Three independent experiments will be needed with average VI of each gene-knocked down cells is obtained. An arbitrary threshold of VI less than 0.5 can be utilized. These groups of genes whose knockdown induced a loss of 50% viability or more were identified as genes of interest, which were considered as reflecting a robust biological effect and then continued with additional validation.
- siRNA validation
Hits identified by a loss of 50% viability of more following siRNA knockdown (VI ≤ 0.5) underwent validation studies. For each hit identified, four different siRNAs (QIAGEN, MD) targeting the same gene were tested in individual wells. Two out of the four siRNAs were the same target sequences as the siRNAs in the screen, when available. The other two siRNAs were new sequences to test. Cells were screened as described above. If at least two out of four of the siRNAs tested reduced viability by at least 50%, the candidate passed validation as a putative hit.
Notes
- We had once screened a total of 44 experimental plates for one RNAi screening experiment. When dealing with multiple plates in one experiment, keep in mind the lipid-siRNA complex formation time should be limited to 10-15 min, record the lipid adding time (CyBio), and calculate the cells dispensing time 10-15 min after (WellMate dispenser). Shorter or longer waiting time can cause variability with transfection efficiency and baseline cell viability, which can be detected using the relevant controls included on each plate.
- As far as setting up the threshold of VI for identifying hits and validation, we chose 0.5 (VI) in our studies. Those genes resulted in a loss of 50% viability or more following gene knockdown were identified as hits. The criteria of choosing an appropriate threshold is that the threshold must reflect a robust biological effect. The value can be adjusted according to projects.
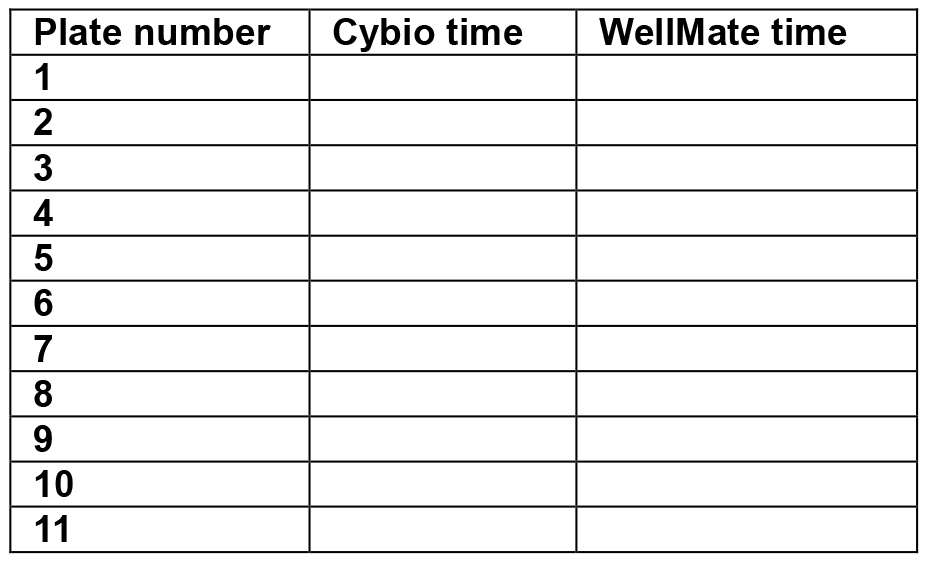
- Time recording
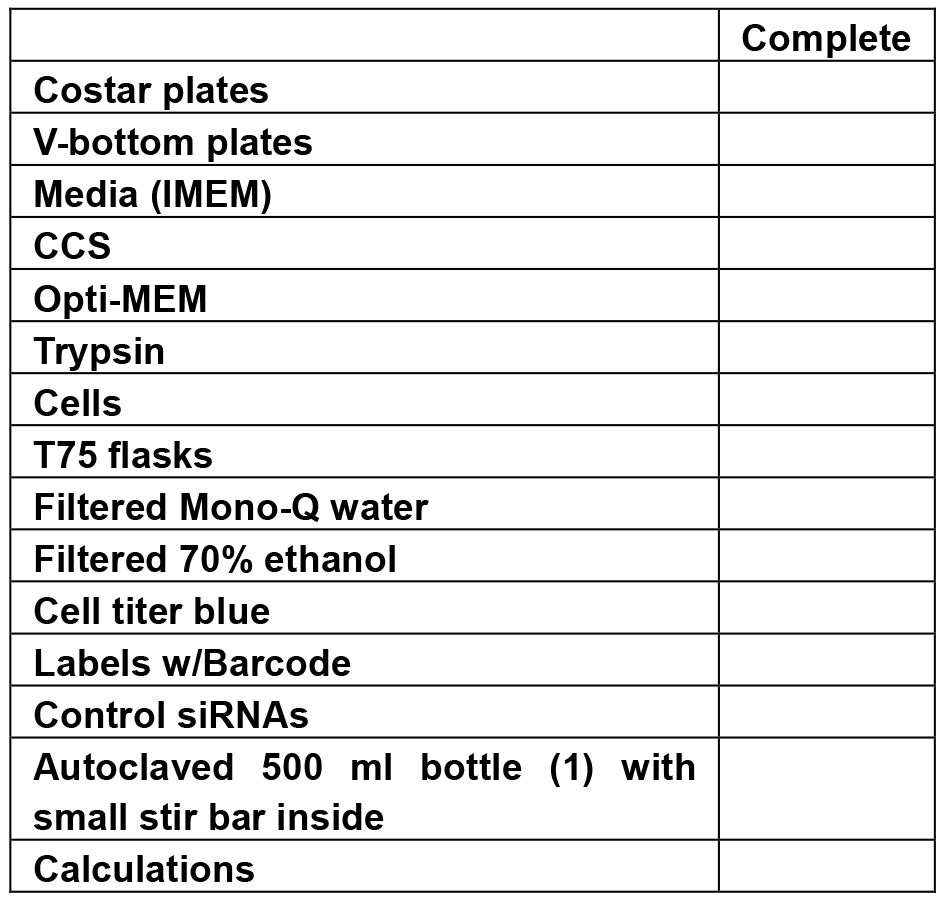
- Screening check list
Acknowledgments
This protocol was adapted from previous work Zhang et al. (2016). We thank Wei Xu, Alan Zwart, David Goldstein and Annie Zuo for their technical assistance. The authors were supported by R01CA050633, CA51880, U54 CA149147 (to LMW), R01CA63366 and R21CA181287 (to EAG).
References
- Astsaturov, I., Ratushny, V., Sukhanova, A., Einarson, M. B., Bagnyukova, T., Zhou, Y., Devarajan, K., Silverman, J. S., Tikhmyanova, N., Skobeleva, N., Pecherskaya, A., Nasto, R. E., Sharma, C., Jablonski, S. A., Serebriiskii, I. G., Weiner, L. M. and Golemis, E. A. (2010). Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci Signal 3(140): ra67.
- Birmingham, A., Selfors, L. M., Forster, T., Wrobel, D., Kennedy, C. J., Shanks, E., Santoyo-Lopez, J., Dunican, D. J., Long, A., Kelleher, D., Smith, Q., Beijersbergen, R. L., Ghazal, P. and Shamu, C. E. (2009). Statistical methods for analysis of high-throughput RNA interference screens. Nat Methods 6(8): 569-575.
- Boutros, M., Ahringer, J. (2008). The art and design of genetic screens: RNA interference. Nat Rev Genet 9(7): 554-566.
- Murray, J.C., Aldeghaither, D., Wang, S., Nasto, R. E., Jablonski, S. A., Tang, Y., Weiner, L. M. (2014). c-Abl modulates tumor cell sensitivity to antibody-dependent cellular cytotoxicity. Cancer Immunol Res 2(12):1186-1198.
- Zhang, J. H., Chung, T. D. and Oldenburg, K. R. (1999). A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4(2): 67-73.
- Zhang, Y. W., Nasto, R. E., Varghese, R., Jablonski, S. A., Serebriiskii, I. G., Surana, R., Calvert, V. S., Bebu, I., Murray, J., Jin, L., Johnson, M., Riggins, R., Ressom, H., Petricoin, E., Clarke, R., Golemis, E. A. and Weiner, L. M. (2016). Acquisition of estrogen independence induces TOB1-related mechanisms supporting breast cancer cell proliferation. Oncogene 35(13): 1643-1656.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Zhang, Y., Nasto, R. E., Jablonski, S. A., Serebriiskii, I. G., Surana, R., Murray, J., Johnson, M., Riggins, R. B., Clarke, R., Golemis, E. A. and Weiner, L. M. (2017). RNA Interference Screening to Identify Proliferation Determinants in Breast Cancer Cells. Bio-protocol 7(15): e2435. DOI: 10.21769/BioProtoc.2435.
Category
Cancer Biology > General technique > Cell biology assays
Cell Biology > Cell-based analysis > Gene expression
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.