- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Using CRISPR/Cas9 for Large Fragment Deletions in Saccharomyces cerevisiae
Published: Vol 7, Iss 14, Jul 20, 2017 DOI: 10.21769/BioProtoc.2415 Views: 14252
Reviewed by: Daan C. SwartsKabin XieAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
CRISPR/Cas9 (Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9) systems have emerged as a powerful tool for genome editing in many organisms. The wide use of CRISPR/Cas9 systems may be due to the fact that these systems contain a simple guide RNA (sgRNA) that is relatively easy to design and they are very versatile with the ability to simultaneously target multiple genes within a cell (Varshney et al., 2015). We have developed a CRISPR/Cas9 system to delete large genomic fragments (exceeding 30 kb) in Saccharomyces cerevisiae. One application of this technology is to study the effects of large-scale deletions of non-essential genes which may give insight into the function of gene clusters within chromosomes at the molecular level. In this protocol, we describe the general procedures for large fragment deletion in S. cerevisiae using CRISPR/Cas9 including: how to design CRISPR arrays and how to construct Cas9-crRNA expression plasmids as well as how to detect mutations introduced by the system within S. cerevisiae cells.
Keywords: CRISPR/Cas9 systemBackground
The CRISPR/Cas9 system is a rapid, efficient, low-cost, and versatile method for genome editing that can be applied in the fields of biology, agriculture, and medicine. To date, several protocols have been reported on how to make large-scale deletions within genomes. Each of these methods contains its own unique characteristics and advantages. The recently developed CRISPR/Cas9 system for excising large stretches of chromosomes has potential advantages over other methods such as the Latour system (Hirashima et al., 2006). The CRISPR/Cas9 system requires two components: (1) the Cas9 endonuclease for DNA cleavage and (2) a variable guide RNA (gRNA) that directs the Cas9 enzyme in a DNA sequence-specific manner (Cong et al., 2013). When Cas9 is targeted to a genomic locus by a gRNA, Cas9 initiates a DSB. The cell will respond to the DSB by repairing the damage via one of two major pathways: high-fidelity homology-dependent repair or error-prone non-homologous end joining (NHEJ).
The CRISPR/Cas9 system described here requires four components: Cas9 endonuclease, CRISPR array, trans-activating crRNA (tracrRNA), and RNase III (this activity is present in the host cell). The CRISPR array is a genomic locus from which pre-crRNAs are transcribed. In this system, the CRISPR array, engineered on the pCRCT plasmid, expresses multiple spacers flanked by direct repeats driven by a single promoter. Cas9 cannot be targeted by crRNA alone, it requires a crRNA-tracrRNA duplex to target it to a specific site within the genome. Two DNA oligonucleotides that encode for spacer sequences interspaced by a direct repeat (DR) were directly synthesized. The formed dsDNA encoding the crRNA was cloned into a Cas9 expression vector. Once the desired plasmid is constructed, transform S. cerevisiae with it and screen the transformants to obtain mutants with large genomic fragment deletions. In this protocol, the deletion efficiency (10%) is lower than described for deletion of genomic fragments using CRISPR/Cas9 in rice (Zhou et al., 2014). There are two possible reasons. The first one is that the genome may be repaired more rapidly in S. cerevisiae in comparison with that in rice. Another one is a stronger selection pressure used for screening rice transformants. In rice, the selection marker for transformants is hygromycin B, while in yeast Uracil as a selection marker is employed. The stronger selection pressure possibly increase the plasmid copy numbers in rice cells.
Materials and Reagents
- Escherichia coli: DH5ɑ (sanyou Biopharmaceuticals)
- S. cerevisiae strain: W303 (MATa ura3-52)
- pCRCT plasmid (Addgene, catalog number: 60621 )
- Salmon DNA (Sigma-Aldrich, catalog number: D1626 )
- Restriction enzyme: 10 U/μl Bsal (Takara Bio)
- 10x buffer G (Takara Bio)
- Taq DNA polymerase (Takara Bio)
- Antibiotic: 100 µg/ml ampicillin (Siyao)
- Pure plasmid mini kit (CWBIO)
- Yeast Gen DNA Kit (CWBIO)
- DNA oligonucleotide primers (GENEWIZ)
- PEG: Poly (ethylene glycol), BioXtra avg. molecular weight 3,350 (Sigma-Aldrich, catalog number: P3640 )
Note: This product has been discontinued.
- Yeast extract (Oxoid, catalog number: LP0021 )
- Tryptone (Oxoid, catalog number: LP0042 )
- Dextrose
- Agar (Solarbio, catalog number: A8190 )
- Sodium chloride (NaCl) (Tianjin Kemiou Chemical Reagent)
- Peptone
- Primers
CasYZF: 5’---ACGCTGTAGAAGTGAAAGTTGG---3’
CasYZR: 5’---TAGTATGCTGTGCTTGGGTGTT---3’
- Glucose (Tianjin Kemiou Chemical Reagent)
- Adenine (Sigma-Aldrich)
- Lithium acetate dihydrate (Sigma-Aldrich, catalog number: L6883-1KG )
- Agarose gel recovery kit (Biomiga)
- YPD liquid media (see Recipes)
- LB plates (with appropriate antibiotics included) (see Recipes)
- YPDA liquid medium (see Recipes)
- SC medium without uracil (see Recipes)
Equipment
- PCR machine (or similar) (Biometra, model: TPfofessional )
- 42 °C water bath (or similar) (XINBAO, catalog number: HH-501BS )
- DNA electrophoresis apparatus (or similar) (SIM International, model: BIO-PRO 200E , catalog number: 0401RHSI049)
- Microcentrifuge (SCILOGEX, model: D2012 )
- Incubator (or similar, capable of incubation of agar plates at 37 °C or 28 °C) (CIMO, model: DNP-III )
Procedure
- Find non-essential S. cerevisiae genes (after deletion or inactivation of which will not result in lethality) with one of the following databases:
http://www.yeastgenome.org/
http://mips.gsf.de/genre/proj/yeast/
http://www.ncbi.nlm.nih.gov/
- Design two 20 nucleotide (nt) spacer sequences for the CRISPR array
Design the two 20 nt spacer sequences for the CRISPR array with the online tool E-CRISP. The following factors should be considered when designing the spacer sequence:
- Spacer sequence length is generally 17-20 nt. The protospacer adjacent motif (PAM) for the Streptococcus pyogenes Cas9 (SpCas9) is 5’-NGG-3’.
- The spacer sequence may be located on the non-transcribed strand or the transcribed strand.
- The selected spacer sequence should be specific to avoid off-target effects.
The tool E-CRISP helps design site-specific spacer sequences.
(http://www.e-crisp.org/E-CRISP/)
- Spacer sequence length is generally 17-20 nt. The protospacer adjacent motif (PAM) for the Streptococcus pyogenes Cas9 (SpCas9) is 5’-NGG-3’.
- Construct the crRNA-Cas9 plasmid
- The dsDNA insertion fragment for the crRNA transcript which contains two spacer sequences and two partial direct repeat sequences has to be synthesized (Figure 1).
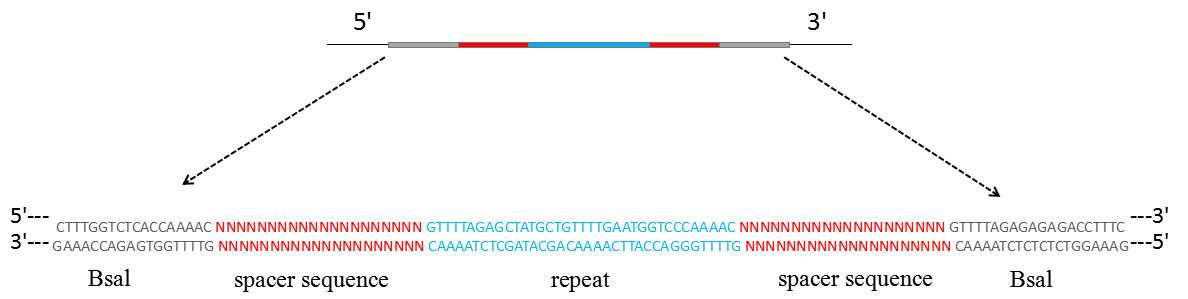
Figure 1. Schematic representation of the dsDNA used for crRNAs expression
- The dsDNA insert for crRNA transcription is digested using BsaI and cloned into the BsaI-digested pCRCT plasmid (Bao et al., 2015).
- DNA digestion reaction:
30 ng of pCRCT plasmid DNA (or the dsDNA)
10x buffer G: 2 µl
Bsal: 1 µl
Add H2O to 20 µl
- Incubate at 37 °C for 3 h.
- Analyze the digestion reaction with 1% (or 2%) (w/v) agarose gel electrophoresis and recover the digested DNA using an agarose gel purification kit.
- Ligate the dsDNA insert for crRNA transcription into pCRCT plasmids
- The ligation reaction:
BsaI digested plasmid: 2 µl (20 ng)
BsaI digested dsDNA insert for crRNA transcription: 0.3 µl (20-50 ng)
Ligation mixture: 2.5 µl
Add H2O to 5 µl
- Incubate at 16 °C overnight.
- Transform E. coli DH5α competent cells with the ligation product.
- Add 5 μl ligation mix into ice-cold competent E. coli DH5α (100 μl).
- Incubate mixture on ice for 30 min and heat-shock it at 42 °C for 90 sec.
- Cool down on ice for 2 min.
- Add 700 μl LB medium (without antibiotics) and incubate at 37 °C and shake at 250 rpm for 45 min.
- Plate 200 μl bacteria suspension on LB plate containing 100 μg/ml ampicillin.
- Incubate overnight at 37 °C in an incubator.
- Run a colony PCR for colonies per transformed pCRCT with CasYZF/CasYZR primers.
PCR protocol
95 °C 5 min 1 cycle
95 °C 30 sec
55 °C 30 sec 30 cycles
72 °C 1 min
72 °C 5 min 1 cycle
4 °C Hold 1 cycle
- Verify the product by sequencing.
- Pick positive clone(s) in 5 ml LB medium with 100 µg/ml ampicillin and incubate for 16 h, at 37 °C, 200 rpm in a shaker.
- Extract plasmids from the transformed DH5α cells using the Pure Plasmid Mini Kit.
- Transform crRNA-Cas9 plasmids into S. cerevisiae competent cells using LiAc/SS carrier DNA/PEG method (Gietz et al., 2007).
- The dsDNA insertion fragment for the crRNA transcript which contains two spacer sequences and two partial direct repeat sequences has to be synthesized (Figure 1).
- Verification of Cas9-crRNA mediated large fragment deletions in S. cerevisiae.
- To verify Cas9-crRNA mediated large fragment deletions, a pair of gene specific primers is required to amplify the targeted region.
- Pick positive clone(s) from selection plate in 5 ml SC medium without uracil and incubate at 28 °C and shake at 200 rpm for 4 d.
- Extract S. cerevisiae genomic DNA from step D2 using Yeast Gen DNA Kit.
- Perform PCR amplification using genomic DNAs extracted from transgenic S. cerevisiae with the following PCR procedure:
Note: P1 and P2 primers (Figure 2, Table 1) are designed based on the sequence flanking the target region to amplify the junction sequence, and amplicon is possible across the ligated junction only when large deletion occurred. P3-P8 primers can confirm the result. The primers can be designed by Primer3 (length: 18-25, GC content: 40%-60%).
PCR protocol
95 °C 5 min 1 cycle
95 °C 30 sec
X °C 30 sec 30 cycles
72 °C N min
72 °C 5 min 1 cycle
4 °C Hold 1 cycle
Note: Annealing temperature (X) and extension time (N) depend on primers.
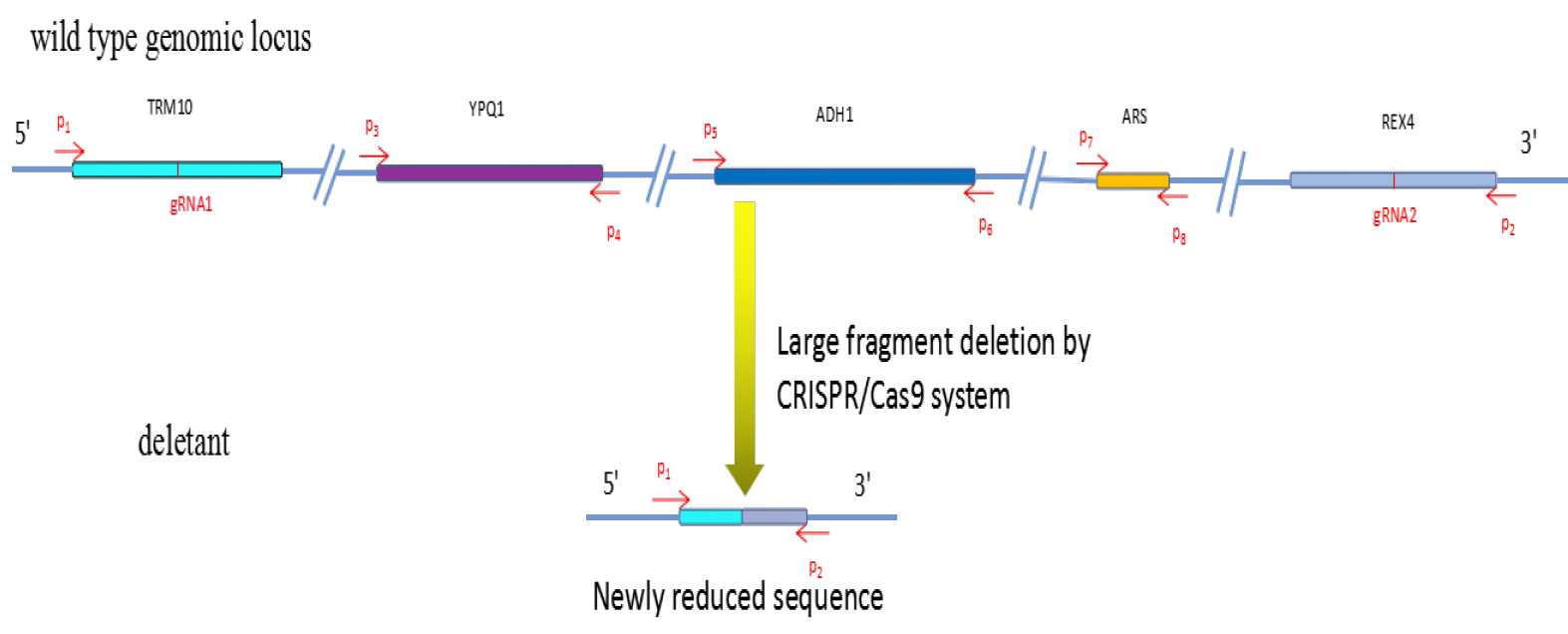
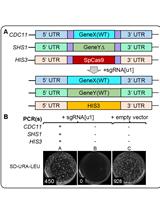
Figure 2. Large fragment deletion by CRISPR/Cas9 as described in this protocol.
See Table 1 for the sequences of P1-P8, gRNA1 and gRNA2
Table 1. The sequences of P1-P8, gRNA1 and gRNA2

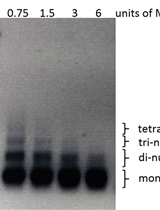
- The PCR product is verified by electrophoresis (Figure 3).

Figure 3. Confirmation of the large chromosomal modifications. A. M, marker; lane 1, mutant; lane 2, control. B, C and D. The PCR product with primers P3-P4, P5-P6, P7-P8 in the target region, respectively, 1a, 1b, 1c, the parental strain. 2a, 2b, 2c, the deletant.
- To verify Cas9-crRNA mediated large fragment deletions, a pair of gene specific primers is required to amplify the targeted region.
Notes
In this protocol, the large-scale deletion efficiency was only 10%. The likely reason for this is that the DSB can be repaired via NHEJ, which typically results in small insertions and/or deletions (indels) at the site of the break. While the HDR pathway allows high fidelity and precise editing, the efficiency of large-scale deletion in S. cerevisiae could be improved if the target vector contains the homology arms of target gene.
Recipes
Note: The solvent for the media is ddH2O.
- YPD liquid media
1% (w/v) yeast extract
2% (w/v) tryptone
2% (w/v) dextrose
Autoclave and store at 4 °C
- LB plates (with appropriate antibiotics included)
1.5% (w/v) agar
1% (w/v) tryptone
0.5% (w/v) yeast extract
1% (w/v) NaCl
Autoclave
- YPDA liquid medium
2% (w/v) peptone
2% (w/v) glucose
1% (w/v) yeast extract
0.005% (w/v) adenine
Autoclave and store at 4 °C
- SC medium without uracil
2% (w/v) glucose
0.67% (w/v) yeast nitrogen base
Autoclave and store at 4 °C
Acknowledgments
This work was supported by the open fund of Key laboratory (No. 3333112) and Bioengineering key discipline of Hebei Province.
References
- Bao, Z., Xiao, H., Liang, J., Zhang, L., Xiong, X., Sun, N., Si, T. and Zhao, H. (2015). Homology-integrated CRISPR-Cas (HI-CRISPR) system for one-step multigene disruption in Saccharomyces cerevisiae. ACS Synth Biol 4(5): 585-594.
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A. and Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819-823.
- Gietz, R. D. and Schiestl, R. H. (2007). Frozen competent yeast cells that can be transformed with high efficiency using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2(1): 1-4.
- Hirashima, K., Iwaki, T., Takegawa, K., Gigahama, Y. and Tohda, H. (2006). A simple and effective chromosome modification method for large-scale deletion of genome sequences and identification of essential genes in fission yeast. Nucleic Acids Res 34(2): e11.
- Varshney, G. K., Pei, W., LaFave, M. C., Idol, J., Xu, L., Gallardo, V., Carrington, B., Bishop, K., Jones, M., Li, M., Harper, U., Huang, S. C., Prakash, A., Chen, W., Sood, R., Ledin, J. and Burgess, S. M. (2015). High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res 25(7): 1030-1042.
- Zhou, H., Liu, B., Weeks, D. P., Spalding, M. H. and Yang, B. (2014). Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice. Nucleic Acids Res 42(17): 10903-10914.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Hao, H., Huang, J., Liu, T., Tang, H. and zhang, L. (2017). Using CRISPR/Cas9 for Large Fragment Deletions in Saccharomyces cerevisiae. Bio-protocol 7(14): e2415. DOI: 10.21769/BioProtoc.2415.
Category
Microbiology > Microbial genetics > Mutagenesis
Microbiology > Microbial genetics > DNA > Chromosomal
Cell Biology > Cell engineering > CRISPR-cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.