- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Assaying Glycogen and Trehalose in Yeast
Published: Vol 7, Iss 13, Jul 5, 2017 DOI: 10.21769/BioProtoc.2371 Views: 10466
Reviewed by: Gal HaimovichAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

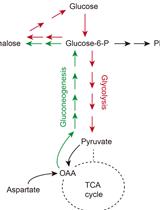

Abstract
Organisms store carbohydrates in several forms. In yeast, carbohydrates are stored in glycogen (a multi-branched polysaccharide) and in trehalose (a disaccharide). As in other organisms, the amount of stored carbohydrate varies dramatically with physiological state, and accordingly, an assay of stored carbohydrate can help reveal physiological state. Here, we describe relatively easy and streamlined assays for glycogen and trehalose in yeast that can be applied either to a few samples, or in a moderately high-throughput fashion (dozens to hundreds of samples).
Keywords: GlycogenBackground
Glycogen and trehalose are the two storage carbohydrates of yeast and many other organisms. In yeast, both these storage carbohydrates accumulate when the medium starts to be depleted and the rate of cell growth decreases. Methods for assaying storage carbohydrates in yeast date back at least to 1956 (Trevelyan and Harrison, 1956a and 1956b), and have been updated many times since (e.g., [Becker, 1978; Quain, 1981; Schulze et al., 1995; Parrou and Francois, 1997; Plata et al., 2013], among others). There are three basic steps in assaying these two storage carbohydrates: first, lysing or permeabilizing the cells; second, freeing glucose from the glycogen or trehalose; and third, assaying the resulting glucose.
Cells can be lysed mechanically (Schulze et al., 1995), but this is inevitably somewhat tedious and time-consuming, and tends to require larger numbers of cells. Cells can be permeabilized by alkali, but glycogen forms large, multi-branched granules, and can be difficult to extract, and so some protocols use both an alkali and an acid extraction (Trevelyan and Harrison, 1956a and 1956b; Quain, 1981). However, alkali treatment alone extracts the vast majority of the glycogen (and probably all of the trehalose) (Becker, 1978; Quain, 1981; Parrou and Francois, 1997); and it may allow enzymes such as amyloglucosidase access to the interior of the permeabilized cell, where it can liberate glucose from any residual glycogen, and alkali extraction alone is much easier than a dual alkali/acid extraction. Therefore, like Becker, and Parrou and Francois, we use only an alkali extraction. However, it is possible that this may fail to assay a relatively small amount of acid-extractable glycogen (Quain, 1981).
In older assays (e.g., [Trevelyan and Harrison, 1956a and 1956b]), glucose was released and/or assayed by purely chemical methods. However, these were relatively non-specific, and also assayed glucose present in other molecules, such as cell wall glucans. Therefore more modern methods use enzymes to liberate glucose from specific polysaccharides; e.g., amyloglucosidases are used to liberate glucose from glycogen (Becker, 1978), and trehalases are used to liberate glucose from trehalose (Parrou and Francois, 1997). A challenge to these methods is that some enzymes are contaminated with other activities. For instance, Parrou and Francois found that some amyloglucosidases were contaminated with trehalases. Therefore either purer enzymes need to be used, or less pure enzymes need to be used under conditions that inhibit the unwanted activities. Here, like Parrou and Francois, we use Aspergillus niger α-amyloglucosidase, which may also contain a trehalase activity (Parrou and Francois, 1997), depending on the specific preparation of enzyme, but we use it at high temperature (55 °C to 57 °C), approximately the optimum temperature for this enzyme, where the trehalase is inactive (Parrou and Francois, 1997).
Finally, the enzymatically-released glucose must be assayed. There are many well-developed assays for glucose. We use the glucose oxidase/peroxidase/o-dianisidine reagent of the Sigma-Aldrich glucose oxidase kit, which produces oxidized o-dianisidine, which has a pink/purple color, easily assayed by absorbance at 540 nm.
Our procedure is adapted from that of Parrou and Francois (1997). However, at most steps, we use smaller volumes of reagents, which make the assay easier in some respects. The small volumes allow us to adapt the procedure to 96-well microtitre dishes, which allows the assay to become moderately high-throughput. We give two procedures, one for 2 ml screw-capped tubes, and one for 96-well microtitre dishes.
Materials and Reagents
- Protective eye wear/safety glasses/face shield
- Pipette tips
- 2 ml screw cap tubes with o-ring (e.g., SARSTEDT, catalog number: 72.694.406 or 72.694.217 )
- Microplate sealing tape (e.g., Corning aluminum tape, Corning, catalog number: 6570 )
- QuickSeal Foil PCR Self Adhesive Seal (Biosero)
Or 4titude PCR Foil Seal (4titude, catalog number: 4ti-0550 )
Or Peelable heat-sealing foil seals and a heat sealer - For 96 well microtitre plate assay
a.Polypropylene, round-bottom 96-well plates, 360 microlitre capacity (e.g., Corning, catalog number: 3359 )
b.Polystyrene, flat-bottom 96-well plates (for plate reader) (e.g., Corning, catalog numbers: 3370 and 3915 ) - Yeast cells
Note: This protocol has been developed for S. cerevisiae. It has not been tried with other species of yeast, but should work. - Milli-Q or double-distilled water
- Glucose assay kit (Sigma-Aldrich, catalog number: GAGO-20 )
- Sulphuric acid
- Glacial acetic acid
- NaAcetate trihydrate*
- Aspergillus niger α-amyloglucosidase (Biochemika, ~70 U/mg) (Sigma-Aldrich, catalog number: 10115 )
Alternatively: 120 U/mg, may be higher purity (Sigma-Aldrich, catalog number: 10113 ). - Porcine trehalase (about 2.3 U/ml) (Sigma-Aldrich, catalog number: T8778 )
- Concentrated H2SO4 (sulfuric acid)*
- Sodium carbonate anhydrous (Na2CO3)*
- 1 M acetic acid (see Recipes)*
- 0.2 M NaAcetate, pH 5.2 (see Recipes)
- 0.2 M NaAcetate, ~pH 8 (see Recipes)
- For 96 well microtitre plate assay
- Concentrated amyloglucosidase buffer (see Recipes)
- Concentrated trehalase buffer (see Recipes)
- Trehalase dilution buffer (0.1 M NaAcetate, pH 5.7) (see Recipes)
- 9 N H2SO4 (see Recipes)
- 0.25 M Na2CO3 (see Recipes)
Note: *Reagents from any qualified company are suitable for this experiment.
Equipment
- Roller or shaker for growing yeast
- Adjustable micropipettes, volumes from 2 to 500 μl
- Spectrophotometer and cuvettes
- Centrifuge (room temperature or chilled) for volumes of 5 to 15 ml
- Microcentrifuge for 1.5 and 2 ml tubes
- Vortex mixer
- pH meter
- Water bath (95 °C)
- Water bath or air incubator, 57 °C and 37 °C
- Glass pipet
- Fume hood
- For 96-well plate assay
- Centrifuge and adaptors for microtitre plates
- Multichannel pipettes
- Plate reader
Note: All those items can be ordered from any qualified company.
Procedure
- Glycogen and trehalose measurements in 2 ml screw-capped tubes
Note: Methods are based on Becker, 1978; Parrou and Francois, 1997.- Cell concentration is measured carefully (e.g., using a Coulter Counter or light scattering in a spectrophotometer) and noted. Transfer samples (of necessarily different, carefully-measured volumes) containing either 5.0 x 107 cells, or, if available, 1.0 x 108 cells, to a 2 ml screw-capped tube and centrifuge at about 20,000 x g for 10 sec. (e.g., 10 sec at maximum speed [~16,000 rpm] at room temperature in an Eppendorf 5415 D).
- Remove the supernatant, wash the cell pellet once in 1 ml ice-cold water to remove residual glucose from the medium. Resuspend cell pellets in 125 μl of 0.25 M Na2CO3 solution with initial vigorous/violent vortexing to thoroughly disperse all cells, and incubate at 95 °C for about 3 h with occasional vortexing (~once per hour, 5 sec), with care to maintain temperature at the top of the tube to avoid excessive condensation.
Note: This can be done by incubating the tubes in an enclosed water bath, so that the air above the tubes is ~95 °C. Somewhat longer or shorter incubations have no apparent effect on the result; Becker (1978) and Quain (1981) incubated for only 90 min. At later times, the alkali-treated cells tend to clump together and are difficult to disperse, but failure to disperse the cells at these late times has no apparent effect on the result. - After incubation at 95 °C, adjust the pH to 5.5, and the volume to 0.5 ml, by addition of 75 μl 1 M acetic acid and 300 μl 0.2 M NaAcetate, pH 5.2 (an appropriate mixture of acetic acid and NaAcetate was made first, and then 375 μl of the mixture was added to the 125 μl of sample).
- Vortex the sample vigorously to resuspend and disperse cell debris, then immediately divide it into two 250 μl in fresh tubes, for glycogen and trehalose measurements respectively.
Note: It may be important to divide the cell debris equally, because the permeabilized cells may still contain glycogen granules, from which glucose will be liberated by amyloglucosidase in the glycogen assay. - For glycogen measurement, make a 20 mg/ml solution of Aspergillus niger α-amyloglucosidase (~70 U/mg) freshly in 0.2 M NaAcetate, pH 5.2. (The pH optimum of the enzyme is ~pH 5, but with high activity between pH 3.0 and pH 6.5 [Pazur and Ando, 1959]). Add 10 μl of this solution to the 250 μl sample. Incubate the mixture overnight at 57 °C, close to the temperature optimum, with occasional vortexing as convenient.
Note: The exact length of the incubation has little apparent effect on the result. However, incubation at temperatures below 55 °C may allow contaminating trehalase activities in the amyloglucosidase to become active, and to release glucose from trehalose, resulting in an over-estimation of the amount of glycogen (Parrou and Francois, 1997). - For trehalose measurement, adjust the pH slightly upwards by addition of 15 μl of 0.2 M NaAcetate (as made by dissolving NaAcetate trihydrate in water, ~pH 8) to the 250 μl sample. The pH optimum for porcine trehalase is about pH 5.8. Add 3 μl of porcine trehalase (2.27 U/ml) (i.e., about 0.007 U of trehalase is added) and mix it well. Incubate the mixture overnight at 37 °C.
- Glucose liberated in the above procedures is quantified using a glucose assay kit.
a.Briefly, prepare the glucose oxidase/peroxidase/o-dianisidine reagent as described by the manufacturer.
b.Centrifuge cell samples for 5 min to pellet cell debris.
c.Mix 50 μl of supernatant from the cell samples with 100 μl of assay reagent, and incubate the mixture in a pre-warmed rack at 37 °C for 30 min.
d.Then add 100 μl of 9 N H2SO4 carefully to each reaction to stop the reaction and develop color (see Recipe 4). - Prepare 50 μl samples of glucose standards (0, 0.5, 1, 1.5, 2, 2.5, 3, 4, 5 μg glucose) in 0.2 M NaAcetate buffer pH 5.2 and assayed in parallel. Typically prepare four or more ‘blank’ (0 glucose) standards. Finally, measure absorbance at 540 nanometers.
- Samples are diluted and re-developed if A540 readings are > 0.5, above which the assay is highly non-linear. A540 readings are converted to absolute amounts of glucose using the standard curve, and normalized for the number of cells used in the sample.
- Cell concentration is measured carefully (e.g., using a Coulter Counter or light scattering in a spectrophotometer) and noted. Transfer samples (of necessarily different, carefully-measured volumes) containing either 5.0 x 107 cells, or, if available, 1.0 x 108 cells, to a 2 ml screw-capped tube and centrifuge at about 20,000 x g for 10 sec. (e.g., 10 sec at maximum speed [~16,000 rpm] at room temperature in an Eppendorf 5415 D).
- Glycogen and trehalose measurements in 96-well microtitre dishes
- For large numbers of assays, the above protocol has been adapted to 96-well microtitre dishes. Cell concentration is measured carefully (e.g., using a Coulter Counter or light scattering in a spectrophotometer) and noted. Transfer samples (of necessarily different, carefully-measured volumes) containing either 5.0 x 107 cells, or, if available, 1.0 x 108 cells, in less than 250 μl of liquid into the wells (~360 μl) of a polypropylene 96-well plate. Centrifuge the plates and aspirate the supernatant. Wash the cell pellets once in 250 μl ice-cold water to remove residual glucose. After centrifugation, remove the supernatant by aspiration.
- Resuspend the cell pellets in 125 μl of 0.25 M Na2CO3 solution. Seal the plates are tightly with a peelable PCR foil microtitre dish sealing film (e.g., QuickSeal Foil PCR Self Adhesive Seal [Biosero] or 4titude PCR Foil Seal) (see Notes). Shake the sealed plates violently by hand, then press to a vortex mixer, to thoroughly disperse all cells.
- Incubate the plates at 95 °C for about 3 h with occasional vortexing, with care to maintain temperature at the top surface of the plate to avoid excessive condensation. This can be done by incubating the plate in an enclosed water bath, so that the air above the plate is ~95 °C.
Note: The polystyrene plates should not be used at this temperature. At later times, the alkali-treated cells tend to clump together and are difficult to disperse, but failure to disperse the cells at these late times has no apparent effect on the result. - After incubation at 95 °C, plates are cooled to room temperature. Immediately before the contents are dispensed into new 96-well plates for treatment with amyloglucosidase or trehalase, shake the plates violently and vortex to disperse cell debris, then tap on the benchtop to bring liquid to the bottom of the wells, then unseal.
- For the glycogen assay, add 188 μl of concentrated amyloglucosidase buffer to each well of a 96-well plate (see Recipes). Pipet 62 μl (half) of the heat-and-alkali treated, freshly-shaken cell suspension into each well. It may be important to divide the cell debris equally, because the permeabilized cells may still contain some glycogen granules, from which glucose will be liberated by amyloglucosidase in the glycogen assay.
- Make a 20 mg/ml solution of Aspergillus niger α-amyloglucosidase (~70 U/mg) freshly in 0.2 M NaAcetate buffer, pH 5.2. Add 10 μl of Aspergillus niger α-amyloglucosidase (~70 U/mg) solution to the 250 μl already in each well (step B3). Seal the plates using ordinary foil seals (e.g., Corning aluminum tape) (see Notes) and shake violently and vortex to mix. Incubate the plates overnight at 57 °C, with occasional vortexing as convenient, with care to maintain high temperature at the top of the plate to prevent condensation (e.g., by placing the entire plate inside a water bath or air incubator.
Note: The exact length of the incubation has little apparent effect on the result. However, incubation at temperatures below 55 °C may allow contaminating trehalase activities in the amyloglucosidase to become active, and to release glucose from trehalose, resulting in an over-estimation of the amount of glycogen (Parrou and Francois, 1997). - For the trehalose assay, add 188 μl of concentrated trehalase buffer (see Recipes) to each well of a 96-well plate. Pipet 62 μl (half) of the heat-and-alkali treated, freshly-shaken cell suspension into each well. Porcine trehalase (2.27 U/ml) is diluted ~3 fold in trehalase dilution buffer (see Materials and Reagents) to 0.7 U/ml, and 10 μl of the diluted enzyme (i.e., about 0.007 U of trehalase) is added to each well. Seal the plate with ordinary foil seals (e.g., Corning aluminum tape) (see Notes), then shake violently and vortex. Incubate the plate overnight at 37 °C.
- Glucose liberated in the above procedures is quantified using a glucose assay kit. Prepare the glucose oxidase/peroxidase/o-dianisidine reagent as described by the manufacturer.
- Add 100 μl of this reagent to each well of a fresh 96-flat-bottom-well, polystyrene plate (i.e., a plate suitable for a plate reader, see Materials and Reagents).
- Centrifuge the 96-well plates containing cell samples for 5 min to pellet cell debris. Pipette 50 μl of supernatant from the cell samples into the 100 μl of assay reagent in the assay plate. Seal the plates and mix by violent shaking and vortexing.
- Incubate the mixture in a water bath at 37 °C for 30 min. Unseal the plates and then add 100 μl of 9 N H2SO4 carefully to each well to stop the reaction and develop color.
Note: Mixing can be accomplished by pipetting up and down; alternatively the plate could be resealed and shaken/vortexed, but with care (e.g., after wrapping in paper towels) because of the sulphuric acid in the wells. - Measure absorbance at 540 nanometers using a plate reader. (see Recipe 4)
- Add 100 μl of this reagent to each well of a fresh 96-flat-bottom-well, polystyrene plate (i.e., a plate suitable for a plate reader, see Materials and Reagents).
- Prepare 50 μl samples of glucose standards (0, 0.5, 1, 1.5, 2, 2.5, 3, 4, 5 μg glucose) in 0.2 M NaAcetate pH 5.2 and assayed in parallel. Typically four or more ‘blank’ (0 glucose) standards are prepared and assayed (Zhao et al., 2016).
- Samples are diluted and re-developed if A540 readings are > 0.5, above which the assay is non-linear. A540 readings are converted to absolute amounts of glucose using the standard curve, and normalized for the number of cells used in the sample.
- For large numbers of assays, the above protocol has been adapted to 96-well microtitre dishes. Cell concentration is measured carefully (e.g., using a Coulter Counter or light scattering in a spectrophotometer) and noted. Transfer samples (of necessarily different, carefully-measured volumes) containing either 5.0 x 107 cells, or, if available, 1.0 x 108 cells, in less than 250 μl of liquid into the wells (~360 μl) of a polypropylene 96-well plate. Centrifuge the plates and aspirate the supernatant. Wash the cell pellets once in 250 μl ice-cold water to remove residual glucose. After centrifugation, remove the supernatant by aspiration.
Data analysis
Linear regression is used to fit a line to the linear part of the standard curve; points in the non-linear range (the higher glucose amounts) are not used in this analysis. The linear regression line is used to convert readings from samples into absolute amounts of glucose. From knowledge of the number of cells in the original sample (here, either 5 x 107 or 1 x 108), and knowledge of the fraction of the sample used in the final color assay (for the 2 ml tube glycogen assay, 50/260 = 0.192), the amount of carbohydrate per cell can be calculated in ng or pg of glucose-equivalents. Two key points for the analysis are accurate knowledge of the number of cells in the starting sample; and use of measurements within the linear range.
Notes
- These assays are highly robust in the sense that the results are not significantly changed by minor or even moderate variation in the incubation times or temperatures, or even the exact volumes of the reagents added. However, at least with some preparations of amyloglucosidase, the incubation temperature must be above 55 °C to avoid contaminating trehalase activity. An indication of amounts of contaminating trehalase activity can be found by using particular preparations of amyloglucosidase trehalose, and then assaying how much glucose is produced.
- We have not asked whether the cell debris remaining after alkali treatment still contains glycogen. This could be done by centrifuging the cell suspension after alkali treatment, and assaying the supernatant and pellet separately for glycogen. If there is little or no glycogen in the pellet fraction, then the protocol could be improved by centrifugation after the alkali step, and use of only the supernatant.
- Maintaining the seal on a 96-well dish for a 3 h incubation at 95 °C can be challenging. A heat sealer with a heat-sealing foil tape provides a robust seal, but heat sealers are expensive and not always available. An adhesive PCR foil tape (e.g., QuickSeal Foil PCR Self Adhesive Seal [Biosero] or 4titude PCR Foil Seal) will suffice if applied carefully. Ordinary foil sealing tape (e.g., Corning) will work if a second, round-bottom 96-well dish with the outer, rectangular flange removed is positioned on top of the foil seal of the sample plate, and the 96 empty wells of the top plate are used to press down, and maintain pressure on, the foil seal of the sample plate. Pressure can be maintained throughout the 3 h incubation with C-clamps, or with a weight. Any particular combination of foil seal, and microtitre dish, should be tested at 95 °C before committing to an experiment.
- A pipetting step could be saved by mixing the enzyme (amyloglucosidase or trehalase) with the buffer before mixing the buffer with the sample. However, we have not tested whether these enzymes are stable at the relatively low pH of these buffers.
- Strains that cannot synthesize glycogen, or that cannot synthesize trehalose (Zhao et al., 2016) can serve as negative controls for the assays. Wild-type cells grown to stationary phase can serve as positive controls.
Recipes
- 1 M acetic acid
Add 1 ml glacial acetic acid (17.4 M) to 16.4 ml ddH2O - 0.2 M NaAcetate buffer, pH 5.2
Dissolve 2.05 g anhydrous NaAcetate in 75 ml of ddH2O in a beaker. While stirring (magnetic stirrer), adjust pH to 5.2 using 1 M acetic acid and a pH meter. Adjust final volume to 100 ml - 0.2 M NaAcetate (~pH 8)
Dissolve 2.05 g anhydrous NaAcetate in 75 ml of ddH2O in a beaker. Adjust final volume to 100 ml - Concentrated amyloglucosidase buffer
Per 96-well plate, mix 3.7 ml of 1 M acetic acid with 15.1 ml of 0.2 M NaAcetate, pH 5.2 - Concentrated trehalase buffer
Per 96-well plate, mix 1.5 ml of 0.2 M NaAcetate (pH ~8), 3.7 ml of 1 M acetic acid, and 15.1 ml of 0.2 M NaAcetate, pH 5.2 - Trehalase dilution buffer (0.1 M NaAcetate, pH 5.7)
Dissolve 1.025 g anhydrous NaAcetate in 75 ml of ddH2O in a beaker. While stirring (magnetic stirrer), adjust pH to 5.7 using 1 M acetic acid and a pH meter. Adjust final volume to 100 ml - 9 N H2SO4
1 vol concentrated H2SO4 diluted to 4 vols final
Note: The preparation of 9 N H2SO4 is highly exothermic and requires care and protective eye and other equipment. It should be done in a fume hood. Add small amounts of the acid, slowly, with stirring, to the water. Have the vessel where the acid and water are mixed pointing away from the experimenter. Wear eye protection. Do not use a polystyrene pipet to add the acid, as the heat generated can be sufficient to melt/deform a polystyrene pipet. - 0.25 M Na2CO3
Dissolve 2.85 g of anhydrous Na2CO3 in 90 ml ddH2O. Make volume up to 100 ml
Acknowledgments
This work was funded by NIH RO1 GM 119175.
References
- Becker, J. U. (1978). A method for glycogen determination in whole yeast cells. Anal Biochem 86(1): 56-64.
- Pazur, J. H. and Ando, T. (1959). The action of an amyloglucosidase of Aspergillus niger on starch and malto-oligosaccharides. J Biol Chem 234(8): 1966-1970.
- Parrou, J. L. and Francois, J. (1997). A simplified procedure for a rapid and reliable assay of both glycogen and trehalose in whole yeast cells. Anal Biochem 248(1): 186-188.
- Plata, M. R., Koch, C., Wechselberger, P., Herwig, C. and Lendl, B. (2013). Determination of carbohydrates present in Saccharomyces cerevisiae using mid-infrared spectroscopy and partial least squares regression. Anal Bioanal Chem 405(25): 8241-8250.
- Quain, D. E. (1981). The determination of glycogen in yeasts. J I Brewing 87: 289-291.
- Schulze, U., Larsen, M. E. and Villadsen, J. (1995). Determination of intracellular trehalose and glycogen in Saccharomyces cerevisiae. Anal Biochem 228(1): 143-149.
- Trevelyan, W. E. and Harrison, J. S. (1956a). Studies on yeast metabolism. 5. The trehalose content of baker's yeast during anaerobic fermentation. Biochem J 62(2): 177-183.
- Trevelyan, W. E. and Harrison, J. S. (1956b). Studies on yeast metabolism. 7. Yeast carbohydrate fractions. Separation from nucleic acid, analysis, and behaviour during anaerobic fermentation. Biochem J 63(1): 23-33.
- Zhao, G., Chen, Y., Carey, L. and Futcher, B. (2016). Cyclin-dependent kinase co-ordinates carbohydrate metabolism and cell cycle in S. cerevisiae. Mol Cell 62(4): 546-557.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Chen, Y. and Futcher, B. (2017). Assaying Glycogen and Trehalose in Yeast. Bio-protocol 7(13): e2371. DOI: 10.21769/BioProtoc.2371.
Category
Microbiology > Microbial metabolism > Carbohydrate
Biochemistry > Carbohydrate > Glycogen
Biochemistry > Carbohydrate > Disaccharide
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.