- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Functional ex-vivo Imaging of Arterial Cellular Recruitment and Lipid Extravasation


Published: Vol 7, Iss 12, Jun 20, 2017 DOI: 10.21769/BioProtoc.2344 Views: 11291
Reviewed by: Gal HaimovichVasiliki KoliarakiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2014

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






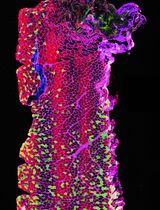
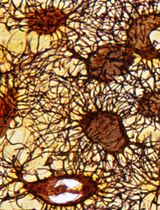
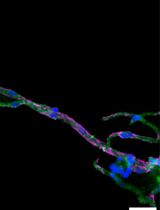
Abstract
The main purpose of this sophisticated and highly versatile method is to visualize and quantify structural vessel wall properties, cellular recruitment, and lipid/dextran extravasation under physiological conditions in living arteries. This will be of interest for a broad range of researchers within the field of inflammation, hypertension, atherosclerosis, and even the pharmaceutical industry. Currently, many researchers are using in vitro techniques to evaluate cellular recruitment, like transwell or flow chamber systems with cultured cells, with unclear physiological comparability. The here introduced method describes in detail the use of a sophisticated and flexible method to study arterial wall properties and leukocyte recruitment in fresh and viable murine carotid arteries ex vivo under arterial flow conditions. This model mimics the in vivo situation and allows the use of cells and arteries isolated from two different donors (for example, wildtype vs. specific knockouts) to be combined into one experiment,thereby providing information on both leukocyte and/or endothelial cell properties of both donors. As such, this model can be considered an alternative for the complicated and invasive in vivo studies, such as parabiotic experiments.
Keywords: ImagingBackground
The core of the method is the application of two-photon laser scanning microscopy (TPLSM) to visualize an ex vivo carotid artery which is mounted in an arteriograph chamber, which has been shown to mimic physiological conditions as present in the in vivo situation (Megens et al., 2007). Fresh arteries, in our case murine carotid arteries but the method is also applicable for other blood vessels of comparable size including human vessels (Bloksgaard et al., 2015), are carefully extracted and mounted in the arteriograph chamber on two glass micropipettes using thin threads. The chamber should provide sufficient space to access the artery with the microscope objective (preferably water dipping or immersion objective with a working distance of > 1 mm). After applying luminal pressure (80 mmHg) and the subsequent correction of the shortening of the length due to isolation (Megens et al., 2007), a variety of fluorescently labeled cells and/or vessel wall components of interest can be perfused into the vessel either under flow or under static conditions. This enables the user to A) count the number of adherent leukocyte subsets, B) determine molecular extravasation (dextrans, lipids) into the arterial wall, C) visualize vascular properties and structures using fluorescently conjugated (specific) antibodies or intrinsic fluorescence signals derived from extracellular matrix components, D) combine the previously described targets.
The general basis of this method was described in 2007 (Megens et al., 2007). Since then this method has been used in several scientific publications, for example to show cell recruitment under control and inflammatory conditions (Schmitt et al., 2014), chemokine presence (Soehnlein et al., 2011), detection of smooth muscle cells (Subramanian et al., 2010; Spronck et al., 2016) or proliferating endothelial cells (Schober et al., 2014), endothelial protein depositions (Ortega-Gomez et al., 2016), adhering platelets (Karshovska et al., 2015), atherosclerotic lesions in the bifurcation (Megens et al., 2007 and 2008; Weber et al., 2011), visualization of the endothelial glycocalyx (Reitsma et al., 2011), or evaluation of extracellular matrix markers (Boerboom et al., 2007; Megens et al., 2007).
TPLSM imaging can be performed prior-, during-, and/or post-perfusion. The settings of the microscope system strongly depend on the available microscope. We utilize a modern Leica SP5II MP system with a 20x WD objective and a Ti:Sa pulsed laser which allows 4 channel imaging at video rate. This is however not a requirement for application of this method as older, less well equipped TPLSM systems also suffice.
In recent years, we have further advanced the method, making it applicable to investigate recruitment of specific cell-types to the viable carotid artery (Döring et al., 2014; Schmitt et al., 2014; Karshovska et al., 2015). Not only does this method enable the user to specifically and simultaneously investigate recruitment of various cell types like monocytes, neutrophils or T-cells to highly physiological endothelium by differential fluorescent labeling (using cell trackers or equivalent), it also allows us to combine specific arteries and cells isolated (blood, bone marrow) from different (wildtype vs. knockout) mouse subsets. As a result, a system is created that can define whether effects on recruitment are mediated by the vascular and/or haematopoietic deficiency. Besides the functional readout of cell recruitment, the method further enables simultaneous or subsequent labelling and subcellular resolution imaging of vascular structures and presence of compounds in the vessel wall. As a result, altered adhesion may directly be linked to the presence or absence of specific targets.
By simultaneous application of various fluorescently labeled leukocyte subsets, the experimental conditions are equal for each cell-type, thereby limiting the experimental variation due to for example flow pattern differences (data may be presented in absolute numbers or ratios). The latter also limits the number of experiments required and, in combination with a reduced number of necessary experimental animals because inflammatory cells and arteries can be isolated from the same animals, ultimately the method reduces the number of required animals.
In addition to the cell recruitment assay we have developed an application using fluorescently labeled low-density lipoprotein particles or dextrans to visualize and quantify lipid or dextran extravasation in viable (diseased) arteries. Lastly, unlike in vivo imaging of large arteries, this ex vivo model does not suffer from unwanted motions of the arterial wall as is the case in the in vivo situation, thereby allowing imaging with subcellular resolution. Moreover, stimuli or specific dyes may be applied at any given time during the experiment giving the researcher full flexibility to tailor the methodology and achieve the required goals.
Materials and Reagents
- Isolation of carotid arteries
- Cell suspension
- 50 ml tubes (3 x) (SARSTEDT, catalog number: 62.547.254 )
- Needle 27 G x 1.5 (Grey) (BD, catalog number: 301629 )
- Syringe 10 ml (1 x) (BD, DiscarditTM catalog number: 309110 )
- Cell strainer 50 µm (Sysmex, CellTrics®, catalog number: 25004-0042-2317 )
- Hanks balanced saline solution (HBSS) with CaCl2 and MgCl2 (Thermo Fisher Scientific, catalog number: 1402550 ), pH 7.4
- Fluorescent cell markers: cell tracker green (Thermo Fisher Scientific, InvitrogenTM, catalog number: C7025 ) and Red (Thermo Fisher Scientific, InvitrogenTM, catalog number: C34565 )
- Ammonium chloride (NH4Cl) (Sigma-Aldrich, catalog number: A9434 )
- Potassium bicarbonate (KHCO3) (Sigma-Aldrich, catalog number: 60339 )
- Ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich, catalog number: E6758 )
- Lysis buffer (see Recipes)
- 50 ml tubes (3 x) (SARSTEDT, catalog number: 62.547.254 )
- Mounting of artery
- Glass etching material (NH4HF2 or NH4F·HF) (Carglass, catalog number: ZB10 EI0003 )
- Needles 14 G x 80 mm, shortened and blunted to 40 mm and 50 mm (Braun Sterican 14 G x 80 mm) (B. Braun Medical, catalog number: 4665473 )
- Nylon thread Ø ~20 µm, for tying of blood vessels (Living Systems Instruments, catalog number: THR-G )
- Three-way tab (2 x) (B. Braun Medical, catalog number: 4095111 )
- Syringe 1 ml (BD, PlastipakTM, catalog number: 300026 )
- Silicone tubing 3 x 1 mm (Carl Roth, catalog number: 9556.1 ) cut to 0.5 m length
- Hanks balanced saline solution (HBSS) with CaCl2 and MgCl2 (Thermo Fisher Scientific, catalog number: 1402550 ), pH 7.4
- Glass etching material (NH4HF2 or NH4F·HF) (Carglass, catalog number: ZB10 EI0003 )
- Molecular extravasation
- Silicone tubing 3 x 1 mm cut to 1.0 m length (Carl Roth, catalog number: 9556.1 )
- Needle 20 G x 1.5 (yellow) (BD, catalog number: 301300 )
- Safe lock tubes 1.5 ml (3 x) (Eppendorf, catalog number: 0030120086 )
- Syringes 1 ml (2 x) (BD, PlastipakTM, catalog number: 300026 )
- Three-way tab (B. Braun Medical, catalog number: 4095111 )
- Fixation tape Durapore 1.25 cm (3M, catalog number: 1538-0 )
- Human Dil-LDL (Kalen Biomedical, catalog number: 770230-9 )
- Hanks balanced saline solution (HBSS) with CaCl2 and MgCl2 (Thermo Fisher Scientific, catalog number: 1402550 ), pH 7.4
- Directly conjugated anti-CD31/eFluor450 (PECAM: Thermo Fisher Scientific, eBioscienceTM, catalog number: 48-0311-82 )
Alternatives: Directly conjugated anti-CD54/A488 (ICAM: BioLegend, catalog number: 116112 ) or anti-CD106/A594 (VCAM: BioLegend, catalog number: 105724 )
- Silicone tubing 3 x 1 mm cut to 1.0 m length (Carl Roth, catalog number: 9556.1 )
- Flow assay
- Silicone tubing 3 x 1 mm cut to 1.5 m and 1.0 m length (Carl Roth, catalog number: 9556.1 )
- Needle 20 G x 1.5 (yellow) (BD, catalog number: 301300 )
- Syringes 1 ml (1 x) (see Reagent C5) and 10 ml (2 x) (BD, DiscarditTM, catalog number: 309110 )
- Three-way Tab (2 x) (B. Braun Medical, catalog number: 4095111 )
- 50 ml tubes (2 x) (SARSTEDT, catalog number: 62.547.254 )
- Fixation tape Durapore 1.25 cm (3M, catalog number: 1538-0 )
- Hanks balanced saline solution (HBSS) with CaCl2 and MgCl2 (Thermo Fisher Scientific, catalog number: 1402550 ), pH 7.4
- Silicone tubing 3 x 1 mm cut to 1.5 m and 1.0 m length (Carl Roth, catalog number: 9556.1 )
Equipment
- Isolation of carotid arteries
- Pen
- FST student spring scissors (Fine Science Tools, catalog number: 91500-09 )
- Dumont forceps (2 x) (Fine Science Tools, catalog number: 91150-20 )
- Scissors (Fine Science Tools, catalog number: 14084-08 )
- Stereomicroscope Leica S8 Apo (LED light source and 0.63x objective) (Leica Microsystems, model: Leica S8 Apo )
- Pen
- Cell suspension
- Timer
- Pen (see Equipment A1)
- Scissors (Fine Science Tools, catalog number: 91401-12 )
- 1 ml pipette (Eppendorf, catalog number: 3120000062 )
- Centrifuge (Eppendorf, model: 5430 )
- Cell counting chamber (Neubauer)
- Cell culture microscope (Leica DMi1 with phase contrast and 10x NA0.3 objective) (Leica Microsystems, model: Leica DMi1 )
- Flow cytometer (BD, model: BD FACSCANTO SYSTEM )
- Timer
- Mounting of artery
- Arteriograph chamber (2 x, IDEE©, Maastricht University, the Netherlands)
- Glass pipettes 1.5 x 0.86 mm (2 x, Harvard Apparatus, catalog number: 30-0057 )
- Pipette puller (NARISHIGE, catalog number: PP-830 )
- Pipette tip grinder (IDEE©, Maastricht University, the Netherlands)
Alternative: Glass pipettes readymade (Living Systems Instruments, catalog number: GCP-300-325 ) - Silicone kit, 73 clear (Farnell, catalog number: 101705 )
- Sphygmomanometer (Riester, Big Ben®, catalog number: 1453-100 ) adapted with a 500 ml air chamber (Schott) and Luer-connectors to fit 1 x 3 mm silicone tubing (IDEE©, Maastricht University, the Netherlands)
- Luer-coupling adapter female (1x) (Carl Roth, catalog number: CT62.1 )
- Stereomicroscope Leica S8 Apo (with LED light source and 0.63x objective) (Leica Microsystems, model: Leica S8 Apo)
- Arteriograph chamber (2 x, IDEE©, Maastricht University, the Netherlands)
- Microscope
- Commercially available Leica SP5IIMP laser scanning microscope system based on a DM6000FS microscope stand (for more info we refer to the manufacturer’s website:
https://www.leica-microsystems.com/products/confocal-microscopes/details/product/leica-tcs-sp5-ii/downloads/)
Alternative: Any functional TPLSM enabling spectral specificity in ≥ 2 detectors and a water dipping objective with sufficient working distance (≥ 1 mm) can be used in combination with the here described assays. - Spectra physics MaiTai DeepSee Ti:Sa laser source
- Leica Hybrid detectors (4 x)
- Luigs and Neumann SM7 motorized microscope stage
- Leica 20x NA1.00WD objective
- Ludin Climate control /laser safety box
- Commercially available Leica SP5IIMP laser scanning microscope system based on a DM6000FS microscope stand (for more info we refer to the manufacturer’s website:
- Molecular extravasation
- Flow assay
- Luer-coupling adapters: male (2 x) (Carl Roth, catalog number: CT58.1 ) and female (1 x) (Carl Roth, catalog number: CT62.1 )
- Syringe infusion pump (Harvard Apparatus pump11 elite) (Harvard Apparatus, catalog number: 70-4504 )
- Column stand with clamp (unknown brand, length 100 cm, footprint 15 x 25 cm)
- Pc with excel and/or paper for cell count
- Luer-coupling adapters: male (2 x) (Carl Roth, catalog number: CT58.1 ) and female (1 x) (Carl Roth, catalog number: CT62.1 )
Software
- Leica LAS AF 2.6 acquisition software
- Leica LAS X 3.11 image processing software (including 3D analyses package); offline
Note: Any processing software package that allows the user to handle multidimensional data can be used.
Procedure
- Isolation of carotid arteries
Isolate the mouse carotid arteries (length after isolation preferably 3-6 mm) following the local ethical committee guidelines using a stereo microscope (see Equipment A5) and place them in HBBS (pH 7.4) in a polystyrene dish (Ø 35 mm) (see Figure 1 for detailed information).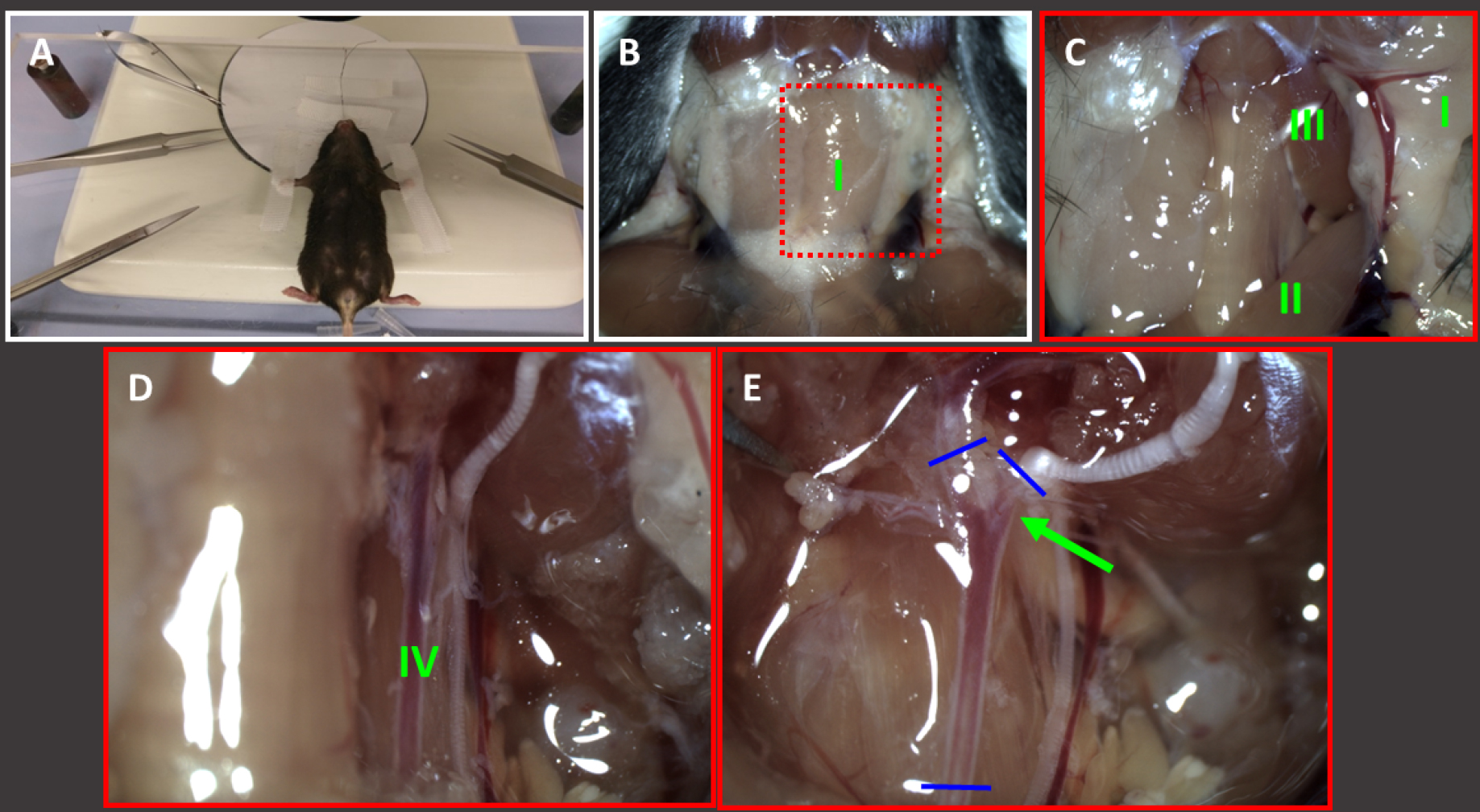
Figure 1. Isolation of carotid arteries. Fix euthanised mouse on the stereomicroscope stage (A) and remove the skin in the neck region using scissors (14084-08) and clean the area using a wet tissue (B). The submaxillary glands (I) become visible. C. Pull one side of the thyrotic gland (I) to the outside and remove musculus sternocleidomastoidius (II) and musculus XXXX (III) by using Dumont forceps. D. Underneath some thin see-through layers, the common carotid artery becomes visible (IV). Carefully remove tissue over the artery and check whether there are atypical sidebranches present. E. Moreover carefully remove tissue from the bifurcation (green arrow) and isolate the cleaned common carotid artery by dissecting it at the blue lines (using spring scissors). Hold the dissected artery by one of both outer end (Dumont forceps) and place it into a small dish filled with HBSS to keep it moist. After isolation the artery must be kept cool (fridge or ice) until further processing.
There are some prerequisites to be met which are crucial for maintaining arterial viability and TPLSM imaging:- Mechanical damage either by pinching or overstretching the vessel must be avoided at all cost as this will immediately result in altered structural and functional behaviour.
- Contact of air with the artery should be minimized. Isolate the artery while keeping the wound moist at all time, after dissection quickly transfer the artery into medium.
- The artery must be very clean for imaging. Any ‘dirt’, hair, or tissue on the outside of the artery will result in loss of visibility of the parts of the artery underneath it. So work meticulously and also remove thin sheets of connective tissue which are covering the artery.
Note: Flushing of the circulation of the mouse, as often used for harvesting of aortas, is not recommended as it potentially introduces air bubbles in the lumen of the arteries which impact the endothelial cells. Flushing can be performed in a controlled way directly after isolation by temporarily mounting (carefully holding it in place with the forceps thus without threads) the common part on an air-free single glass pipette in the perfusion chamber and gently flushing HBSS through it. This however has to be done quickly (within 30 min) after carotid isolation in order to avoid blood clots! (See also procedure in section ‘mounting of artery’.)
- Mechanical damage either by pinching or overstretching the vessel must be avoided at all cost as this will immediately result in altered structural and functional behaviour.
- Isolation of bone marrow derived leukocytes
- Isolate the femur and/or tibia of the mice following the local ethical committee guidelines and place them in HBBS (pH 7.4).
Note: The most convenient method of isolation is to first remove the skin from the hind limbs, subsequently pull on the hind limbs to dislocate the hip. Afterwards it is easy to remove the hind limb by cutting in the hip area, without the risk of cutting the bone. After removal, remaining muscles and tissue can be safely removed. - Cut off the epiphyses (heads) of the bones with scissors so that the marrow cavity is open.
- Flush the bone with HBSS into a 50 ml tube by using a 27 G needle and 10 ml syringe.
Note: It is important that all bone marrow cells are removed from the bones. Use as much volume as needed for flushing until practically all cells (red colour) are removed. - Prepare a single cell solution by resuspending the cells using a 1 ml pipette.
- Filter the resuspended bone marrow cells with a 50 µm cell strainer to reduce tissue in the final cell suspension.
- Centrifuge the cells for 5 min at 300 x g.
- Resuspend the pellet with 5 ml of lysis buffer (in order to lyse the red blood cells) and incubate for 1 min at room temperature.
- Centrifuge the cells for 5 min at 300 x g.
Note: If required, different leukocyte subsets (e.g., monocytes, neutrophils and lymphocytes) can be isolated according to lab specific protocols. - Prepare cell tracker solution by diluting the cell tracker 1:1,000 in HBSS.
Note: Different colours of cell tracker can be used to distinguish between leukocyte subsets (e.g., monocytes, neutrophils and lymphocytes) or between different leukocyte sources (e.g., wild-type vs. knock-out) - Resuspend the cells in prepared cell tracker solution (total volume 3 ml per leukocyte subset/source).
- Incubate for 15 min at room temperature.
Notes: - Alternate cell tracker green and cell tracker red between the different experimental runs to avoid any impact of the dyes on the results as well as to keep the observers blinded.
- Other cell marker combinations such as Calcein (eBioscience) and Rhodamin (Sigma-Aldrich) or other cell tracker combinations may also be used.
- Wash the cells in suspension twice after the staining procedure, resuspend in 3 ml HBSS, use 50 ml tubes, centrifuge at 300 x g for 5 min.
Note: Washing of the cells is critical in order to avoid overall staining of the vessel wall by unbound cell trackers in the prepared cell suspension which causes loss of contrast! - Count total numbers of cells per leukocyte subset/source using a Neubauer chamber and a microscope (with phase contrast).
- Mix both types of leukocytes (2 different subsets or sources, labelled with different colours to distinguish them from each other) to achieve a 1:1 ratio of both resuspended in a total of 6 ml HBSS.
- Cell density for each leukocyte group should be 1.5 x 106 leukocytes (thus, the total number of cells in suspension for each flow assay is 3 x 106 in 6 ml HBSS).
- Analyse the resulting leukocyte suspension by flow cytometry in order to confirm the 1:1 ratio.
- Isolate the femur and/or tibia of the mice following the local ethical committee guidelines and place them in HBBS (pH 7.4).
- Preparation of arteriography chamber
- Pull and grind pipettes from 0.58 x 1 x 80 mm glass tubes by using a pipette puller (heater = 60; weight= 3 x large block) and a grinder setup. The goal is to generate pipettes with a length of 40 mm and 50 mm and a tip diameter of 250-350 µm with a 45° tip angle. Tip size and angle can be checked using any bright field microscope (≈ 100x magnification) and a scale bar slide (see Figure 2).

Figure 2. Micropipette construct. A. Overview picture micropipette as used for home-built perfusions chamber. The glass micropipette is glued into a blunted and shortened 14 G needle (green, total length approximately 50 mm). B. The glass pipette sticks approximately 0.8-10 mm out of the blunted needle. C. Close up of the tip of the pipette which has a tip diameter of 250-300 µm and is grinded to obtain a 45° angle. After gluing and grinding the tip is etched in order to create a roughened glass surface (not visible) giving better friction for the mounting of the artery. - Etch the tips of the grinded pipettes by NH4HF2 or NH4F·HF compound to the tip of the grinded pipettes (one can simply hold the tip several mm in the compound in the opened storage flask). (10-20 min or when the etching process was successful), rinse carefully with distilled water after the procedure, and let them dry (RT). Etched tips enable more stable tying of the artery to the glass.
Note: Because of differences in arteriography chamber design the preparation and properties of the pipettes are different for the various chambers. The most important property of the pipette for this method is the tip opening diameter and the 45° angular shape of the tip since both directly influence the flow pattern within the artery. - Cut the 14 G needles by making a circular edge using a small file and then breaking the tip off using small pincers and remove any sharp edges using sandpaper. The needles should have a length of 40 mm and 50 mm (50 mm for the adjuster side of the chamber).
- Glue the glass pipettes into the shortened and blunt 20 G needles using silicone kit. Make sure the space between the glass and the steel needle is fully closed to ensure air tightness of the construct. Moreover, make sure the non-sharpened outer edge of the glass pipette just reaches the Luer-lock connector of the 20 G needle in order to avoid accumulation of debris in the non-filled space within the construct.
- Carefully rinse the created constructs with distilled water after the procedure and store them safely or mount them in the arteriography chambers.
- Pull and grind pipettes from 0.58 x 1 x 80 mm glass tubes by using a pipette puller (heater = 60; weight= 3 x large block) and a grinder setup. The goal is to generate pipettes with a length of 40 mm and 50 mm and a tip diameter of 250-350 µm with a 45° tip angle. Tip size and angle can be checked using any bright field microscope (≈ 100x magnification) and a scale bar slide (see Figure 2).
- Mounting of artery
- Prepare a thread dish by applying some tape (5 cm) upside down (sticky side up) in a polystyrene Petri dish (Ø 100 mm). Tape the outer ends of the upside down tape to the polystyrene dish with some short pieces of tape.
- Prepare threads (Ø ~20 µm; living systems instrumentation) in the thread dish (Figure 3) using a stereomicroscope (see Equipment A5). The threads are generated by 1) curve the straight thread and make a circle by crossing the outer ends with each other, 2) leashing one outer end twice through the circle.

Figure 3. Preparation of the nylon threads as used for mounting of an artery. A. Overview of materials needed for the procedure; 10 cm polystyrine Petri dish (I) with Durapore tape and 8-10 mm long nylon threads (Ø ≈ 20 µm: arrow); 2 Dumont forceps (II), and the nylon thread (III). B-F. Close ups of Durapore tape with nylon threads showing the stepwise procedure of thread preparation; B. Single thread next to a cluster of threads (green arrow); C. Curve the straight thread and make a circle by crossing the outer ends with each other; D. Placing one outer end in the circle (diameter 500-700 µm) using the sticky properties of the tape to keep it in place; E. Leashing one outer end through the circle; F. By repeating Procedure D and Procedure E the thread is ready. Place the finished thread with its circle half on the sticky Durapore and half over the tape for improved handling (threads are visible and gradable without the use of a stereomicroscope). - Clean the interior and the pipettes of the home built (but also commercially available) arterial perfusion chamber (Figure 4A) carefully with 70% ethanol (1x flush) and HBSS (2x flush), attach a 3-way tab on both pipettes with the female Luer-couplers and fill with HBSS medium with CaCl2 and MgCl2 (pH 7.4). Make sure that both pipettes are completely air-free and leave a 1 ml syringe filled with medium connected to the 3-way tab at the side of the length adjuster of the chamber.
- Transfer the freshly isolated and cleaned carotid artery to the perfusion chamber and place it at the stereomicroscope (see Equipment A5) (see Figure 4).

Figure 4. Mounting of murine carotid artery in home-built perfusion chamber. A. Example of a perfusion chamber with longitudinal length adjuster (green arrow); B. Fill the clean perfusion chamber with HBSS and attach 3-way tabs (I) on both pipettes. Remove any air from both pipettes using a HBSS filled 1 ml syringe (II). C. View of the interior of the perfusion chamber using a stereomicroscope; D. Close up of a glass pipette with 2 pre-prepared nylon threads slid over it; E. Overview of mounted but unpressurized artery. Two threads are used on each pipette. The bifurcation (III) helps determining the correct flow direction. F. The mounted artery is then closed at the flow outlet (bifurcation side) and connected to a sphygmomanometer (IV) at the flow entrance side to reveal any leakage of the mounted artery. G. Stepwise increase pressure to and correct the distance between both pipettes (using the adjuster, green arrow) until the artery is straight at 80 mmHg. Note the length difference between the artery in Procedure E and Procedure G, where a pressurized artery at 80 mmHg is approximately 30% longer. - Place two or three threads over the pipette using the stereomicroscope (see Equipment A5).
- Mount the common part of the carotid by pulling it over the pipette at the length adjuster side of the chamber (Not too far!).
- Tighten the two or three threads so that the artery is fixed in between the glass tip and the threads.
- Gently flush some medium through the carotid in order to get rid of possible blood leftovers.
Note: It is preferred to flush the artery inside the chamber and not flush the mouse as a total in order to limit air in the lumen. This however has to be done quickly (within 30 min) after carotid isolation in order to avoid blood clots! - Adjust the distance between both pipettes using the adjuster on the outside of the chamber (Figure 4B; green).
- Place two or three threads over the 2nd pipette.
- Mount the bifurcation side of the carotid by pulling it over the pipette (Figure 4B). Make sure you have tightened all branches that are not cannulated to create a closed circuit.
Notes: Make sure you mount the artery in its physiological direction for the flow assay, i.e., flow goes from common to bifurcation. A failure of the flow direction will result in false-positive or negative results. Also make sure the artery is not mounted under tension due to twisting of the artery! - Open the 3-way tab on the exit side (the non-length adjustment side of the chamber) and gently flush some medium to see whether pipettes are open (HBSS runs through the artery and comes out on the exit side) and artery is mounted stably.
- Connect the sphygmomanometer with 3 x 1 mm silicone tubing and a female Luer-connector to the length adjustment side of the chamber and fill the 3 x 1 mm tubing of the manometer with some HBSS.
- Adjust the 3-way connectors to create a closed circuit that can be pressure controlled using the manometer and stepwise increase the average intraluminal pressure while adjusting the length of the unfolding vessel using the external adjuster until you reach 100 mmHg. Be aware of leaks of the mounted artery by tracking the medium level in the 3 x 1 mm silicone tube connected to the sphygmomanometer.
Note: A leak somewhere in the total system will result in alteration of the buffer level in the 3 x 1 mm silicone tubing! - Make sure the length is corrected properly at 100 mmHg and then lower the pressure to 80 mmHg (mimicking physiological stretch of the artery).
- When all is correctly mounted, proceed to the next step.
- Prepare a thread dish by applying some tape (5 cm) upside down (sticky side up) in a polystyrene Petri dish (Ø 100 mm). Tape the outer ends of the upside down tape to the polystyrene dish with some short pieces of tape.
- Two-photon laser scanning microscopy (TPLSM)
- Set the climate box to 37 °C. Make sure the temperature sensor inside the climate box is positioned close to the objective. To reach a stable temperature the climate control box must be started approx. 1 h prior to the first measurement. No additional CO2/O2 gas control is required for these experiments.
- Turn on the system according to protocol (different for every microscope).
- Place the perfusion chamber onto the microscope stage and focus on the carotid artery using the 20x 1.00 WD objective, the fluorescent light source and the RGB-filter.
Note: Be very careful not to focus too deep as this will result in destruction of the sample, chamber and potentially the objective! - Select the spectral channels required (channel one: 400-430 nm for collagen; 440-500 nm for elastin; 510-540 nm for green cell tracker; 620-670 nm for cell tracker deep red).
- Tune the excitation laser (Ti:Sa) to 800 nm.
- Select the imaging mode (XYZ), scan speed (200-400 Hz), and xyz resolution (XY-pixel format according to Nyquist criterion, Z-step 0.8-1.5 µm). For creating movies of the artery during flow/cell adhesion, choose XYT or XYZT scans, select ‘minimize’ time interval and maximize overall recording time. Moreover, reduce the number of pixels and increase scan speed to 600 Hz in order to have a reasonable refreshment rate of the pictures.
- Select the area of interest in the mounted carotid artery in ‘live’ mode; avoid the first field of view directly next to the glass pipettes as this area suffers from mechanical damage due to the mounting protocol!
Note: The easiest way to find the vessel is to focus on the threads that tighten the artery to the pipette. The threads will appear very bright (autofluorescence) and by looking at the loop one can derive whether focus is on the top or the lower part of the artery. - Select the Z-range (total thickness of the imaged volume).
- Start the selected scan using the ‘record’ function.
- Record as many datasets as required.
- When ready with imaging make sure to rename and save the generated data.
- Copy the data to one of the processing computers.
- Shut down according to the TPLSM protocol. Don’t forget the fluorescent light bulb and the climate chamber!
Note: The here described TPLSM settings should be considered as an example only. Settings of the TPLSM system strongly depend on the layout of the system and as such should be optimized specifically for every different system! Try to achieve maximum detection efficiency with lowest bleed through for the required channels (minimum of 2 channels, preferably 3-4 channels).
- Set the climate box to 37 °C. Make sure the temperature sensor inside the climate box is positioned close to the objective. To reach a stable temperature the climate control box must be started approx. 1 h prior to the first measurement. No additional CO2/O2 gas control is required for these experiments.
- Static experiment for LDL permeability (Figure 5)
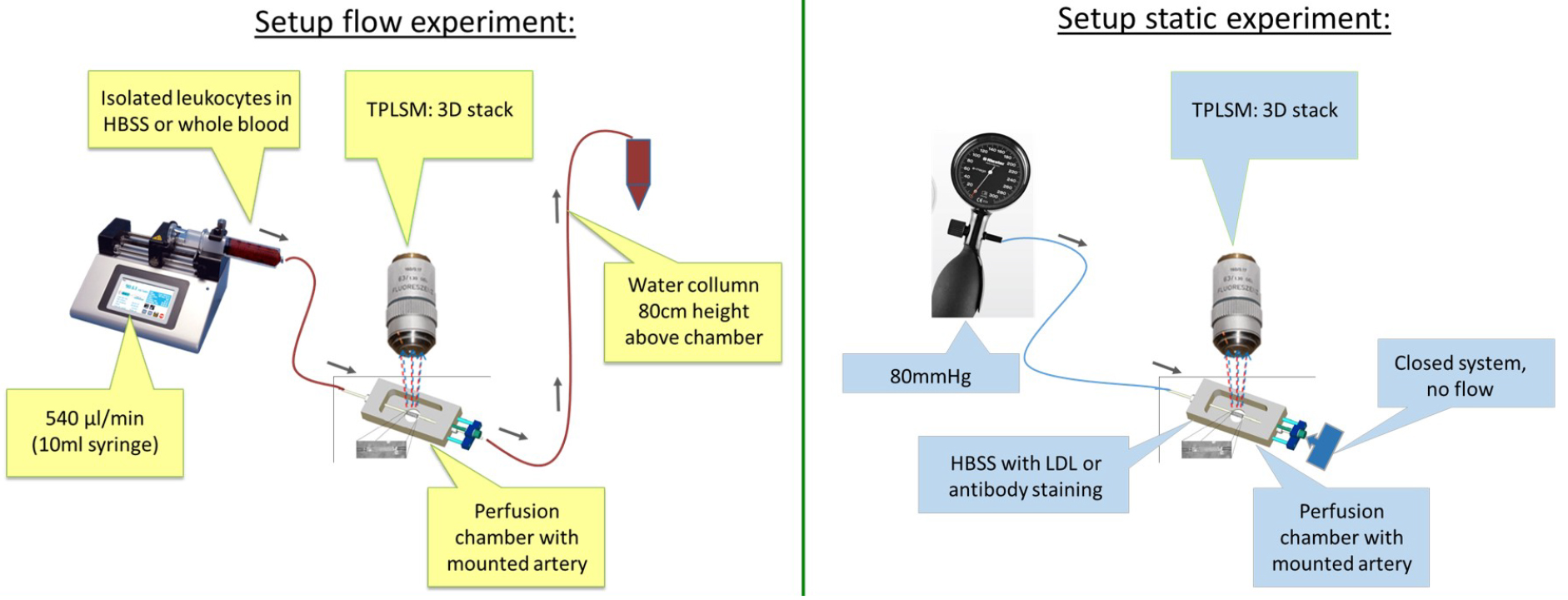
Figure 5. Schematic overview of experimental setup of flow experiment and static experiment- After the mounting procedure, reduce the luminal pressure again and open the 3-way tab on the exit side.
- Remove the 3-way connector on the inlet side.
- Remove any air from the inlet Luer-connector (using a 1 ml syringe filled with HBSS with a 14 G needle) of the pipette.
- Slowly inject mounted vessels with 50 µg/ml Dil-LDL, diluted in HBSS using a 1 ml syringe.
Note: DiI is a fluorescent lipophilic tracer attached to LDL with an excitation of 549 nm and emission of 565 nm. - Re-pressurize the artery to 80 mmHg using the sphygmomanometer.
- Place the sphygmomanometer outside on top of the climate box and keep an eye on it while imaging. A drop in pressure means that the system is not completely closed and therefore leaky.
- Place the mounted artery on the microscope stage inside the climate box.
- Incubate for 90 min at 37 °C.
- Perfuse the vessel with HBSS to wash away non-bound LDL (similar procedure as previously described injection of Dil-LDL).
- Inject anti-CD31 eFluor450 antibody (labeling endothelial cells, more specifically endothelial junctions): same procedure as previously described injection of Dil-LDL.
- Incubate for 10 min at 37 °C.
- Perfuse the vessel with HBSS to wash away non-bound antibody (similar procedure as previously described injection of Dil-LDL), re-pressurize and place it again on the microscope stage.
Note: Flushing or loading of the vessel is best performed outside the climate chamber with help of a stereomicroscope. - Start recording (see image acquisition section above).
- When analyzing permeability, one needs to scan the whole artery systematically! Keep the field-of-view (f.o.v.) size and scanned volume equal and move field by field through the artery until you have reached the other pipette.
Note: Discard the last f.o.v. closest to the pipettes because of potential damage due to the mounting procedure. - Quantification of LDL uptake is performed after the experiments in recorded xyz-scans using processing software (Figure 6).
Alternative: Any image processing software that enables working with multidimensional datasets will suffice.
Note: Approximately 25% of the overall artery can be visualized from one side when using f.o.v. of 450 x 450 µm and when scanning from tunica adventitia to the lumen until both vessel wall borders disappear from the field of view (see Figure 6C for an impression of the scanned volume). In case a total overview of the artery is required, one can turn the carotid artery mechanically with 90°, 180° and 270° by simultaneous turning of both pipettes (careful!). It is not advised to focus directly through the artery to reach the ‘bottom’ because this will strongly reduce the image quality and increase risk of focusing too deep/destroying the sample.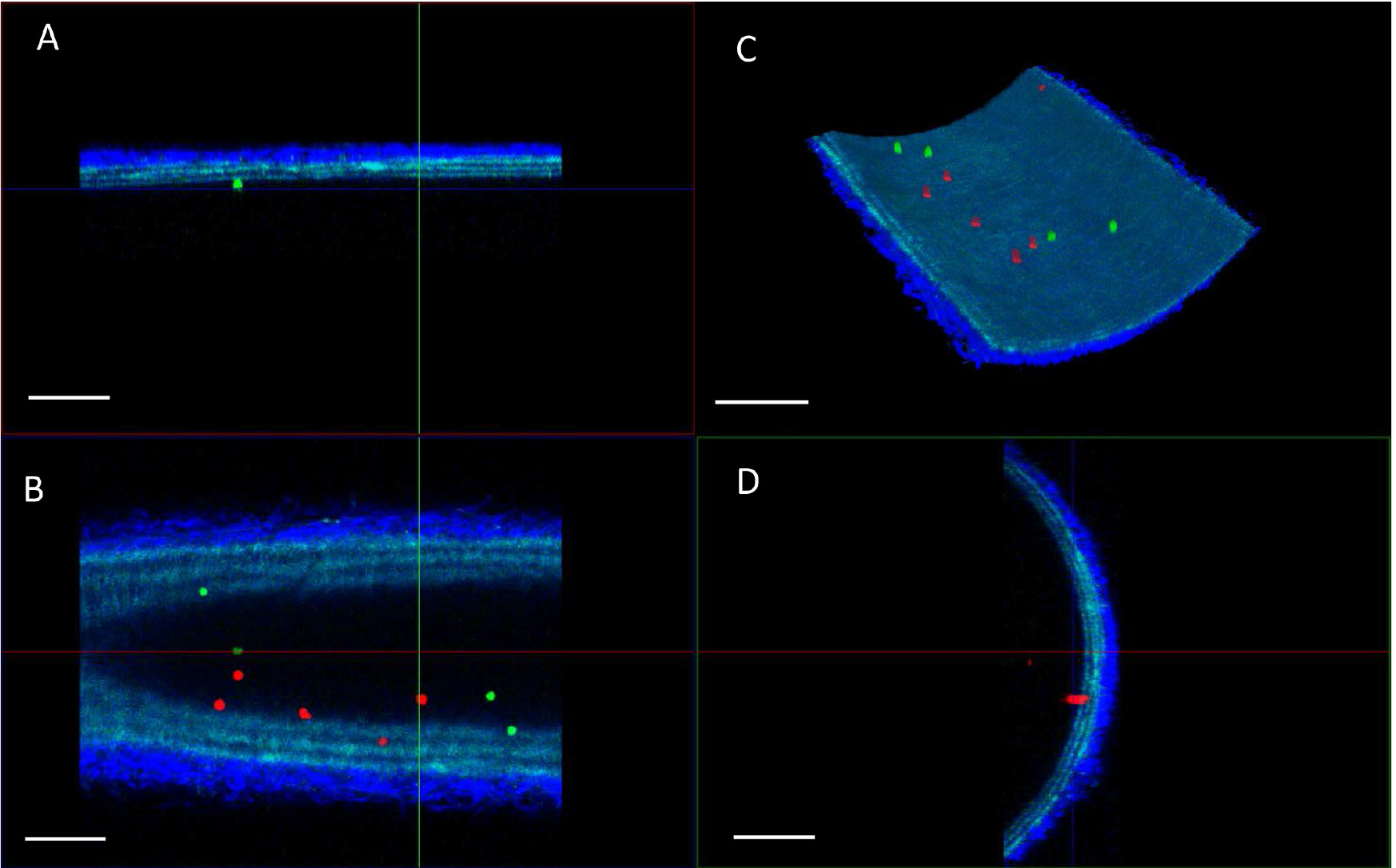
Figure 6. Examples of TPLSM imaging of bone marrow derived leukocyte adhesion of two classes (green; wildtype leukocytes, red; knock out leukocytes) in carotid artery after the flow assay. A. XZ projection; B. XY projection; C. 3D reconstruction, and D. YZ projection of a single field-of-view. The arterial wall extracellular matrix components collagen (bright blue, tunica adventitia) and elastin (blue, tunica media) are visualized using the intrinsic signals of autofluorescence and second harmonics generation. Scale bars = 100 µm.
- After the mounting procedure, reduce the luminal pressure again and open the 3-way tab on the exit side.
- Dynamic (flow) experiment for cell recruitment (Figure 5)
- Connect the flow chamber with the common carotid artery side to the syringe pump using a 10 ml syringe, a male Luer-connector, a 3-way tab, 3 x 1 mm silicone tubing (1 m), and a female Luer-connector (from the syringe to the 3-way connector attached to the chamber). The pump can be placed on top of or next to the climate chamber and should be tilted slightly to make sure the syringe empties completely. Use some extra length of 3 x 1 mm silicone tubing (1 m) and make a loop inside the climate chamber (with durapore tape) in order to preheat the medium or cell suspension to 37 °C before it reaches the artery.
- Make sure the whole system is air-free and filled with HBSS before attaching it to the artery!
- Connect the exit side of the vessel/chamber to the water column using another 3-way tab and 3 x 1 mm silicone tubing. Make sure the water column is filled with HBSS and positioned at the correct height (black line). Use a 50 ml tube attached to the clamp and the column at 80 cm height above the position of the artery to collect the ‘flushed’ fluids. Tape the 3 x 1 mm silicone tubing to the falcon to stabilize the position of the silicone tubing going into the 50 ml tube.
- Select the appropriate flow rate on the pump (see manual). Make sure you select the correct syringe (size and brand), flow direction (push) and total volume (5.5 ml, dependent on the experiment).
- For mimicking arterial flow rate set the pump to 0.54 ml/min (calculated based on flow rates and average carotid diameters in vivo).
- Open the 3-way tabs in the appropriate way and perform a test run with medium only to check the open flow system. Fluid should slowly drip out of the 3 x 1 mm silicone tubing into the 50 ml tube on the water column.
- Change the syringe with a similar one containing the pre-prepared cell suspension mix. Make the Luer-connector and 3-way tab air free using a 14 G needle and an HBSS filled 1 ml syringe. Apply a total volume of 6 ml of cell suspension mix, which allows for a flow time of 10 min.
Note: Make sure that every time you change syringes there are no air bubbles introduced to the system as air bubbles will alter or destroy the endothelial cells of the arteries! Air bubbles may be avoided by carefully removing air/filling the outer end of Leur-connectors with HBSS (using a 1 ml syringe with HBSS and a 20 G needle) before (re)connecting syringes or 3-way tabs. Also make sure that the silicone tubing is properly filled with HBSS before attaching it to the chamber. - When image acquisition settings are set, start the flow procedure.
- When a live recording of the flow assay is needed, start the xyzt scan.
Note: Adhesive cell will be round-shaped and flowing cells in the lumen will appear as stripes due to the laser scanning methodology of the TPLSM. - Once the cell suspension has run out, replace the cell suspension syringe with the HBSS containing syringe and start the flow again for 3 min (or approximately 2 ml). The latter is to avoid cell loss in the dead volume of the silicone tubing on the pump side and to reduce the number of non-adhesive cells in the luminal space.
Alternative: One could add specific antibodies to the washing flush of HBSS to visualize additional targets or structures of interest. - For still xyz-images (Figure 6), stop the pump.
- When analyzing cell recruitment, one needs to scan the whole artery systematically! Keep the field of view (f.o.v.) size and scanned volume equal and move field by field through the artery until you’ve reached the other pipette (discard the f.o.v.’s closest to the pipettes).
- Counting of adhesive cells can be performed live (while scanning through the artery; 2 observers are needed and always record some reference xyz-scans) or after the experiments in recorded xyz-scans (Figure 6) using processing software (Leica LasX)
Notes: - Approximately 25% of the overall artery can be visualized from one side when using f.o.v. of 450 x 450 µm and when scanning from tunica adventitia to the lumen until both vessel wall borders disappear from the field of view (see Figure 6C for an impression of the scanned volume). In case a total overview of the artery is required, one can turn the carotid artery mechanically with 90°, 180° and 270° by simultaneous turning of both pipettes (careful!). It is not advised to focus directly through the artery to reach the ‘bottom’ because this will strongly reduce the image quality and increase risk of focusing too deep/destroying the sample.
- Optimally, the cell count is performed by two blinded observers.
- Required time is approximately 2.5 h per artery: from isolation of the artery and preparation of the cell suspension, up to counting of adhesive cells on 50% of the endothelial surface. In case detailed imaging of additional targets is required, another 30-60 min should be added to the total time. As a result, one may realistically perform 4 flow experiments per working day. Arteries can be stored on ice or in a fridge for a while (up to 4 h did not change the live/dead staining pattern; Megens et al., 2007) but in order to improve reproducibility of the experiments, it is advisable to process them as quickly as possible after isolation.
- Repeat the whole procedure for the carotid artery derived from the 2nd donor (either wildtype or knock-out mouse) mounted in a 2nd arteriography chamber.
Note: To reduce the influence of experimental timing and vessel chamber properties it is best to set up the next round of experiments with alternating frequency (i.e., use the 2nd mouse type as the first one in the initially used chamber) and alternate the applied cell markers for staining the isolated leukocytes subsets (i.e., green becomes red in the next series of experiments).
- Connect the flow chamber with the common carotid artery side to the syringe pump using a 10 ml syringe, a male Luer-connector, a 3-way tab, 3 x 1 mm silicone tubing (1 m), and a female Luer-connector (from the syringe to the 3-way connector attached to the chamber). The pump can be placed on top of or next to the climate chamber and should be tilted slightly to make sure the syringe empties completely. Use some extra length of 3 x 1 mm silicone tubing (1 m) and make a loop inside the climate chamber (with durapore tape) in order to preheat the medium or cell suspension to 37 °C before it reaches the artery.
Data analysis
- Analyses of Dil-LDL extravasation
- Quantification of the number and size of junctional Dil-LDL particles per cell was performed using Leica LASX 3.11 software.
Note: Any image processing software that enables multidimensional analyses will be useable for this method. - Recorded xyz-stacks were recombined into maximum intensity projections, transforming the 3D information into a resultant 2D image showing all cell junction in the xy-plane (Figure 7A).
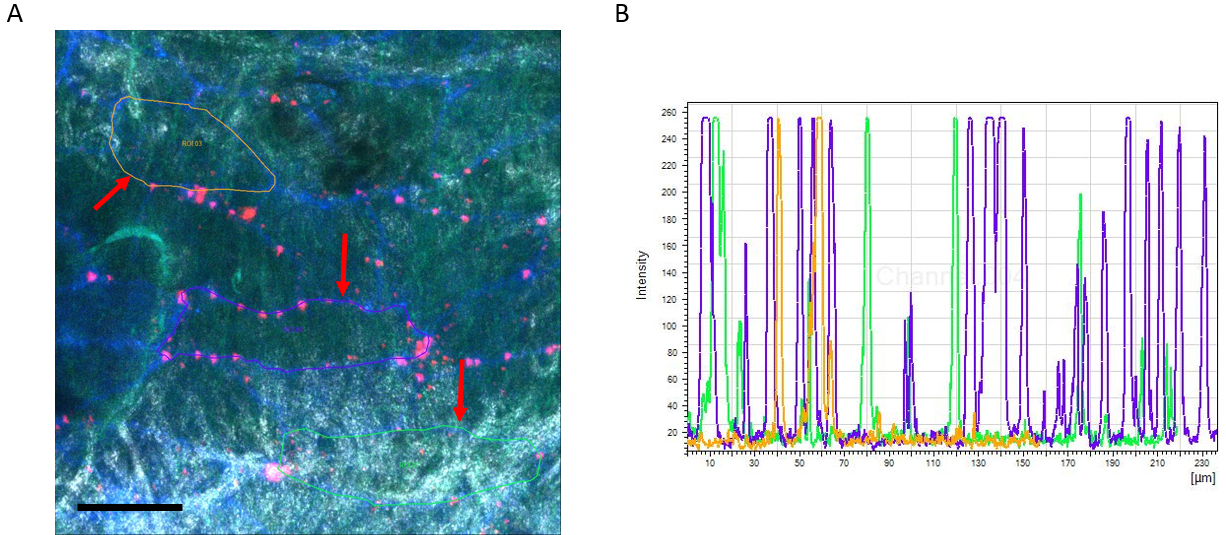
Figure 7. Example of TPLSM imaging of Dil-LDL extravasation in the carotid artery. A. Max. image showing XY projection (Dil-LDL in Red and CD31 in Blue). The arterial wall extracellular matrix components collagen (white, tunica adventitia) and elastin (green, tunica media) are visualized using the intrinsic signals of autofluorescence and second harmonics generation. Image also includes analysis method showing selection of ROIs (red arrows). Scale bar = 50 µm. B. Example of intensity scheme showing individual peaks representing the Dil-LDL particles. - A further reduction of back ground noise was achieved using a blur filter (kernel size 3).
- Ten individual endothelial cells were chosen based on the junctional CD31 signal and blinded from the LDL signal by temporarily excluding the spectral channel representing the Dil-LDL particles.
- A polyline was drawn around each selected cell based on its CD31 junctional staining, resulting in specific regions of interest for each individual cell (Figure 7A, red arrows).
- The polyline selection was used to generate intensity profiles (Figure 7B).
- A threshold of the Dil-LDL channel was set just above the background intensity to exclude background noise.
Note: The specific threshold level will vary depending on the experimental background level and has to be determined individually. - The total number of individual peaks in the intensity profile represented the number of particles, the full width of each peak was used to estimate the size of junctional Dil-LDL particles.
Note: Dead cells were excluded and the cells used for the quantification did not share any cellular junctions.
- Quantification of the number and size of junctional Dil-LDL particles per cell was performed using Leica LASX 3.11 software.
Recipes
- Lysis buffer
155 mM NH4Cl
10 mM KHCO3
0.1mM EDTA
Dissolved in 1 L water (MilliQ), pH 7.2
Acknowledgments
The study was financially supported by the DFG (SFB1123 TB Z01, A01 and B03 and INST409/97-1 FUGG) and the Alexander von Humboldt Foundation. Christian Weber is supported by the ERC.
References
- Bloksgaard, M., Leurgans, T. M., Nissen, I., Jensen, P. S., Hansen, M. L., Brewer, J. R., Bagatolli, L. A., Marcussen, N., Irmukhamedov, A., Rasmussen, L. M. and De Mey, J. G. (2015). Elastin organization in pig and cardiovascular disease patients' pericardial resistance arteries. J Vasc Res 52(1): 1-11.
- Boerboom, R. A., Krahn, K. N., Megens, R. T., van Zandvoort, M. A., Merkx, M. and Bouten, C. V. (2007). High resolution imaging of collagen organisation and synthesis using a versatile collagen specific probe. J Struct Biol 159(3): 392-399.
- Döring, Y., Noels, H., Mandl, M., Kramp, B., Neideck, C., Lievens, D., Drechsler, M., Megens, R. T., Tilstam, P. V., Langer, M., Hartwig, H., Theelen, W., Marth, J. D., Sperandio, M., Soehnlein, O. and Weber, C. (2014). Deficiency of the sialyltransferase St3Gal4 reduces Ccl5-mediated myeloid cell recruitment and arrest: short communication. Circ Res 114(6): 976-981.
- Karshovska, E., Zhao, Z., Blanchet, X., Schmitt, M. M., Bidzhekov, K., Soehnlein, O., von Hundelshausen, P., Mattheij, N. J., Cosemans, J. M., Megens, R. T., Koeppel, T. A., Schober, A., Hackeng, T. M., Weber, C. and Koenen, R. R. (2015). Hyperreactivity of junctional adhesion molecule A-deficient platelets accelerates atherosclerosis in hyperlipidemic mice. Circ Res 116(4): 587-599.
- Megens, R. T., Oude Egbrink, M. G., Cleutjens, J. P., Kuijpers, M. J., Schiffers, P. H., Merkx, M., Slaaf, D. W. and van Zandvoort, M. A. (2007). Imaging collagen in intact viable healthy and atherosclerotic arteries using fluorescently labeled CNA35 and two-photon laser scanning microscopy. Mol Imaging 6(4): 247-260.
- Megens, R. T., Oude Egbrink, M. G., Merkx, M., Slaaf, D. W. and van Zandvoort, M. A. (2008). Two-photon microscopy on vital carotid arteries: imaging the relationship between collagen and inflammatory cells in atherosclerotic plaques. J Biomed Opt 13(4): 044022.
- Megens, R. T., Reitsma, S., Schiffers, P. H., Hilgers, R. H., De Mey, J. G., Slaaf, D. W., oude Egbrink, M. G. and van Zandvoort, M. A. (2007). Two-photon microscopy of vital murine elastic and muscular arteries. Combined structural and functional imaging with subcellular resolution. J Vasc Res 44(2): 87-98.
- Ortega-Gomez, A., Salvermoser, M., Rossaint, J., Pick, R., Brauner, J., Lemnitzer, P., Tilgner, J., de Jong, R. J., Megens, R. T., Jamasbi, J., Doring, Y., Pham, C. T., Scheiermann, C., Siess, W., Drechsler, M., Weber, C., Grommes, J., Zarbock, A., Walzog, B. and Soehnlein, O. (2016). Cathepsin G controls arterial but not venular myeloid cell recruitment. Circulation 134(16): 1176-88.
- Reitsma, S., Oude Egbrink, M. G., Heijnen, V. V., Megens, R. T., Engels, W., Vink, H., Slaaf, D. W. and van Zandvoort, M. A. (2011). Endothelial glycocalyx thickness and platelet-vessel wall interactions during atherogenesis. Thromb Haemost 106(5): 939-946.
- Schmitt, M. M., Megens, R. T., Zernecke, A., Bidzhekov, K., van den Akker, M. N., Rademakers, T., van Zandvoort, M. A., Hackeng, T. M., Koenen, R. R. and Weber, C. (2014). Endothelial JAM-A guides monocytes into flow-dependent predilection sites of atherosclerosis. Circulation 129(1): 66-76.
- Schober, A., Nazari-Jahantigh, M., Wei, Y., Zhe, Z., Gremse, F., Grommes, J., Megens, R. T. A., Heyll, K., Thiemann, A., Iruela-Arispe, M. L., Wang, S., Kiessling, F., Olson, E. N. and Weber, C. (2014). MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat Med 20(4): 368-376.
- Soehnlein, O., Wantha, S., Simsekyilmaz, S., Doring, Y., Megens, R. T., Mause, S. F., Drechsler, M., Smeets, R., Weinandy, S., Schreiber, F., Gries, T., Jockenhoevel, S., Moller, M., Vijayan, S., van Zandvoort, M. A., Agerberth, B., Pham, C. T., Gallo, R. L., Hackeng, T. M., Liehn, E. A., Zernecke, A., Klee, D. and Weber, C. (2011). Neutrophil-derived cathelicidin protects from neointimal hyperplasia. Sci Transl Med 3(103): 103ra198.
- Spronck, B., Megens, R. T., Reesink, K. D. and Delhaas, T. (2016). A method for three-dimensional quantification of vascular smooth muscle orientation: application in viable murine carotid arteries. Biomech Model Mechanobiol 15(2): 419-432.
- Subramanian, P., Karshovska, E., Reinhard, P., Megens, R. T., Zhou, Z., Akhtar, S., Schumann, U., Li, X., van Zandvoort, M., Ludin, C., Weber, C. and Schober, A. (2010). Lysophosphatidic acid receptors LPA1 and LPA3 promote CXCL12-mediated smooth muscle progenitor cell recruitment in neointima formation. Circ Res 107(1): 96-105.
- Weber, C., Meiler, S., Doring, Y., Koch, M., Drechsler, M., Megens, R. T., Rowinska, Z., Bidzhekov, K., Fecher, C., Ribechini, E., van Zandvoort, M. A., Binder, C. J., Jelinek, I., Hristov, M., Boon, L., Jung, S., Korn, T., Lutz, M. B., Forster, I., Zenke, M., Hieronymus, T., Junt, T. and Zernecke, A. (2011). CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J Clin Invest 121(7): 2898-2910.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
van der Vorst, E. P., Maas, S. L., Ortega-Gomez, A., Hameleers, J. M., Bianchini, M., Asare, Y., Soehnlein, O., Döring, Y., Weber, C. and Megens, R. T. (2017). Functional ex-vivo Imaging of Arterial Cellular Recruitment and Lipid Extravasation. Bio-protocol 7(12): e2344. DOI: 10.21769/BioProtoc.2344.
Category
Cell Biology > Tissue analysis > Tissue isolation
Immunology > Immune cell imaging > Two-photon microscopy
Cell Biology > Tissue analysis > Tissue imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.