- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Protocol for Production of Mutant Mice Using Chemically Synthesized crRNA/tracrRNA with Cas9 Nickase and FokI-dCas9
Published: Vol 7, Iss 11, Jun 5, 2017 DOI: 10.21769/BioProtoc.2340 Views: 11743
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system is the most widely used genome editing tool. A common CRISPR/Cas9 system consists of two components: a single-guide RNA (sgRNA) and Cas9. Both components are required for the introduction of a double-strand break (DSB) at a specific target sequence. One drawback of this system is that the production of sgRNA in the laboratory is laborious since it requires cloning of an sgRNA sequence, in vitro transcription reaction and sgRNA purification. An alternative to targeting Cas9 activity by sgRNA is to target it with two small RNAs: CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA). Both of these small RNAs can be chemically synthesized which makes the production of these RNAs less difficult when compared to sgRNA. Another downside of the CRISPR/Cas9 systems is that off-target effects have been reported. However, modified forms of Cas9 have been developed to minimize off-target effects. For example, nickase-type Cas9 (nCas9) and FokI domain-fused catalytically-inactive Cas9 (FokI-dCas9; fCas9) induce DSBs only when two guide RNAs bind opposite strands within a defined distance. In this protocol, we describe our experimental system for the production of mutant mice using a CRISPR/Cas9 system that combines crRNA, tracrRNA, and modified forms of Cas9. This method not only facilitates the preparation of reagents for the genome editing system but it can also reduce the risk of off-target effects.
Keywords: CRISPR/Cas9Background
The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system is an effective genome editing tool. In bacteria, CRISPR/Cas9 functions as an adaptive immune system. It consists of two small RNAs, CRISPR RNA (crRNA) and trans-activation crRNA (tracrRNA) and the Cas9 DNA nuclease, which digests targeted DNA (Jinek et al., 2013). Several groups have established the CRISPR/Cas9 system as a tool for introducing mutations in many cell types (Cong et al., 2013; Mali et al., 2013). When the Cas9 nuclease is targeted to genomic DNA, it cleaves DNA resulting in a lesion that is repaired by non-homologous end joining (NHEJ) or homologous DNA recombination. Since NHEJ can be an error-prone mechanism, mutations can be introduced into the genome when DNA is repaired by this mechanism. The CRISPR/Cas9 system can be used to edit the genomes of mice by microinjecting Cas9 and the single-guide RNA (sgRNA) into fertilized eggs. Although sgRNAs have been used extensively with success, the generation of the sgRNA is laborious because the sgRNA must be cloned from DNA oligomers and then transcribed in vitro. Systems that use crRNA and tracrRNA can eliminate much of the labor involved in preparing sgRNAs since crRNA and tracrRNA are small enough in length to be chemically synthesized. So instead of injecting Cas9 with sgRNA, mutant mice can be obtained by microinjecting Cas9 with both crRNA and tracrRNA (crRNA/tracrRNA). One drawback of using the CRISPR/Cas9, is guide RNA/Cas9 complex can generate off-target mutations, which are unintended mutations that occur at loci with similar sequences to the target sequence of the guide RNA. The risk of inducing off-target mutations can be reduced by incorporating the use of modified forms of Cas9, such as the nickase-type of Cas9 (nCas9) and the FokI domain-fused catalytically-inactive Cas9 (FokI-dCas9; fCas9). These Cas9 forms cleave target DNA only when two sgRNAs bind opposite strands with a limited distance between them (Ran et al., 2013; Hara et al., 2015). Recently, we successfully generated mutant mice by microinjecting modified Cas9s with chemically synthesized crRNA/tracrRNA into fertilized eggs (Terao et al., 2016). This protocol reduces time and labor for the preparation of targeting RNA and can reduce the risk of off-target effects.
Materials and Reagents
- 35 mm dish*
- 60 mm dish*
- Mouth pipettes*
- 0.22 µm filter*
- Microloader (Eppendorf, catalog number: 5242956003 )
- Glass capillary for mouth pipettes (Drummond Scientific, catalog number: 1-000-0500 )
- Glass capillary for holding pipettes (Sutter Instrument, catalog number: B100-75-10 )
- Glass capillary for injection pipettes (World Precision Instruments, catalog number: TW100F-4 )
- Male and female C57BL/6 x DBA/2 hybrid (B6D2F1) or other strains (for recipient zygotes)
- Male and female ICR (for preparation of pseudopregnant mice)
- Cas9 plasmids (available from Addgene)
- Nickase-type of Cas9 (nCas9) (Addgene, catalog number: 41816 )
- FokI domain-fused catalytically-inactive (fCas9) (Addgene, catalog number: 52970 )
- PrimeSTAR MAX (Takara Bio, catalog number: R045A ) or other PCR polymerases
- Qiaquick PCR Purification Kit (QIAGEN, catalog number: 28104 ) or equivalent
- AgeI*
- mMESSAGE/mMACHINE T7 Transcription Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM1344 )
- MEGAclear Transcription Clean-Up Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM1908 )
- Pregnant mare serum gonadotropin (PMSG) (ASKA Animal Health, Serotropin®, catalog number: 879412 )
- Human chronic gonadotropin (hCG) (ASKA Animal Health, catalog number: Gonatropin 3000 )
- Hyaluronidase (Sigma-Aldrich, catalog number: H4272 )
- KSOM medium (ARK resource)
- Dichlorodimethylsilane (Tokyo Chemical Industry, catalog number: D0358 )
- ExoSAP-IT (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 78200.200.UL )
- Sodium chloride (NaCl) (Wako Pure Chemical Industries, catalog number: 191-01665 )
- Potassium chloride (KCl) (Nacalai Tesque, catalog number: 28514-75 )
- Calcium chloride (CaCl2·2H2O) (Sigma-Aldrich, catalog number: C7902 )
- Potassium phosphate monobasic (KH2PO4) (Sigma-Aldrich, catalog number: P5655 )
- Magnesium sulfate (MgSO4·7H2O) (Sigma-Aldrich, catalog number: M2773 )
- Sodium bicarbonate (NaHCO3) (Sigma-Aldrich, catalog number: S5761 )
- HEPES (DOJINDO, catalog number: 342-01375 )
- Sodium DL-lactate (Sigma-Aldrich, catalog number: L7900 )
- Sodium pyruvate (Sigma-Aldrich, catalog number: P2256 )
- D-(+)-glucose (Sigma-Aldrich, catalog number: G7528 )
- Polyvinyl alcohol (Sigma-Aldrich, catalog number: P8136 )
- Penicillin-streptomycin (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 )
- Phenol red (Sigma-Aldrich, catalog number: P0290 )
- Sodium hydroxide (NaOH) (Sigma-Aldrich)
- Paraffin liquid (Nacalai Tesque, catalog number: 26137-85 )
- BIOTAQ (Bioline, catalog number: BIO-21040 ) or other PCR polymerases
- M2 medium (see Recipes)
*Note: No particular preference.
Equipment
- Spectrophotometer (Thermo Fisher Scientific, model: NanoDrop ND-1000 )
- Capillary puller (Sutter Instrument, model: P-1000 )
- Microforge (NARISHIGE, model: MF-900 )
- Micro centrifuge*
- Thermal cycler*
- Heating block*
- Upright microscope*
- Micromanipulator (NARISHIGE, model: NT-88-V3 )
- Injectors (NARISHIGE, models: IM-11-2 and IM-9B )
- FemtoJet (Eppendorf, model: FemtoJet® Express )
- CO2 incubator (37 °C, 5% CO2, and 95% air condition)
*Note: No particular preference.
Procedure
- crRNA design
- To select target sequences of crRNAs, a list of possible sgRNAs are required first. We recommend using online web tools such as CRISPR design tool (http://crispr.mit.edu/) or CRISPRdirect (https://crispr.dbcls.jp/) for this purpose. Choose a pair of sgRNAs with the following criteria: spacer distance is 4-20 base pairs (bp) for nCas9 or is 14-19 bp for fCas9 (Figure 1) and the pair of sgRNAs must be located on opposite strands of genomic DNA (Ran et al., 2013; Hara et al., 2015).
- Design crRNA sequences by removing the protospacer adjacent motif (PAM) and replacing T (thymine) with U (uracil) from the sequences of a pair of sgRNAs as shown in Figure 1.
- Have selected crRNAs (42 nt) and tracrRNA (69 nt) chemically synthesized and purified by HPLC. We adjust the concentration of crRNAs and tracrRNA to 1 µg/µl.

Figure 1. Design of crRNA target sites for modified Cas9s. (Top) crRNA target sites are underlined in genomic DNA. Both the sense and antisense strands of a portion of the genomic sequence at the intronic region of the Bcr gene are shown. (Middle) The two target sites for crRNAs are underlined. Protospacer adjacent motif (PAM) sequences (NGG) appear in bold letters. The spacer sequence is indicated by a gray dashed double-headed arrow. (Bottom) crRNA sequence (42 nt) for each target site and the sequence of tracrRNA (69 nt) are shown. The crRNA-specific sequence (22 nt), derived from Jinek et al. (2013), is shown in blue. Uracil is shown in red.
- To select target sequences of crRNAs, a list of possible sgRNAs are required first. We recommend using online web tools such as CRISPR design tool (http://crispr.mit.edu/) or CRISPRdirect (https://crispr.dbcls.jp/) for this purpose. Choose a pair of sgRNAs with the following criteria: spacer distance is 4-20 base pairs (bp) for nCas9 or is 14-19 bp for fCas9 (Figure 1) and the pair of sgRNAs must be located on opposite strands of genomic DNA (Ran et al., 2013; Hara et al., 2015).
- Preparation of Cas9 mRNA
- Preparation of DNA templates for in vitro transcription
- Synthesize Cas9 mRNA in an in vitro transcription reaction using T7 RNA polymerase. Note, the plasmid that contains fCas9 has a T7 promoter sequence, but the plasmid that contains nCas9 does not (Figure 2). To add the T7 promoter sequence to the 5’ terminal of nCas9, amplify nCas9 by PCR with the following primers:
T7-Cas9-F: TAATACGACTCACTATAGGGAGAATGGACAAGAAGTACTCCATTGG (underline: T7 promoter sequence)
Cas9-R: TCACACCTTCCTCTTCTTC
Check the PCR product (4,163 bp) by agarose gel electrophoresis. - Purify PCR products with the Qiaquick PCR Purification Kit (according to the manufacturer’s instructions). The concentration of purified products can be determined using a spectrophotometer.
- Digest the plasmid containing fCas9 with AgeI and purify the products with the Qiaquick PCR Purification Kit (according to the manufacturer’s instructions).
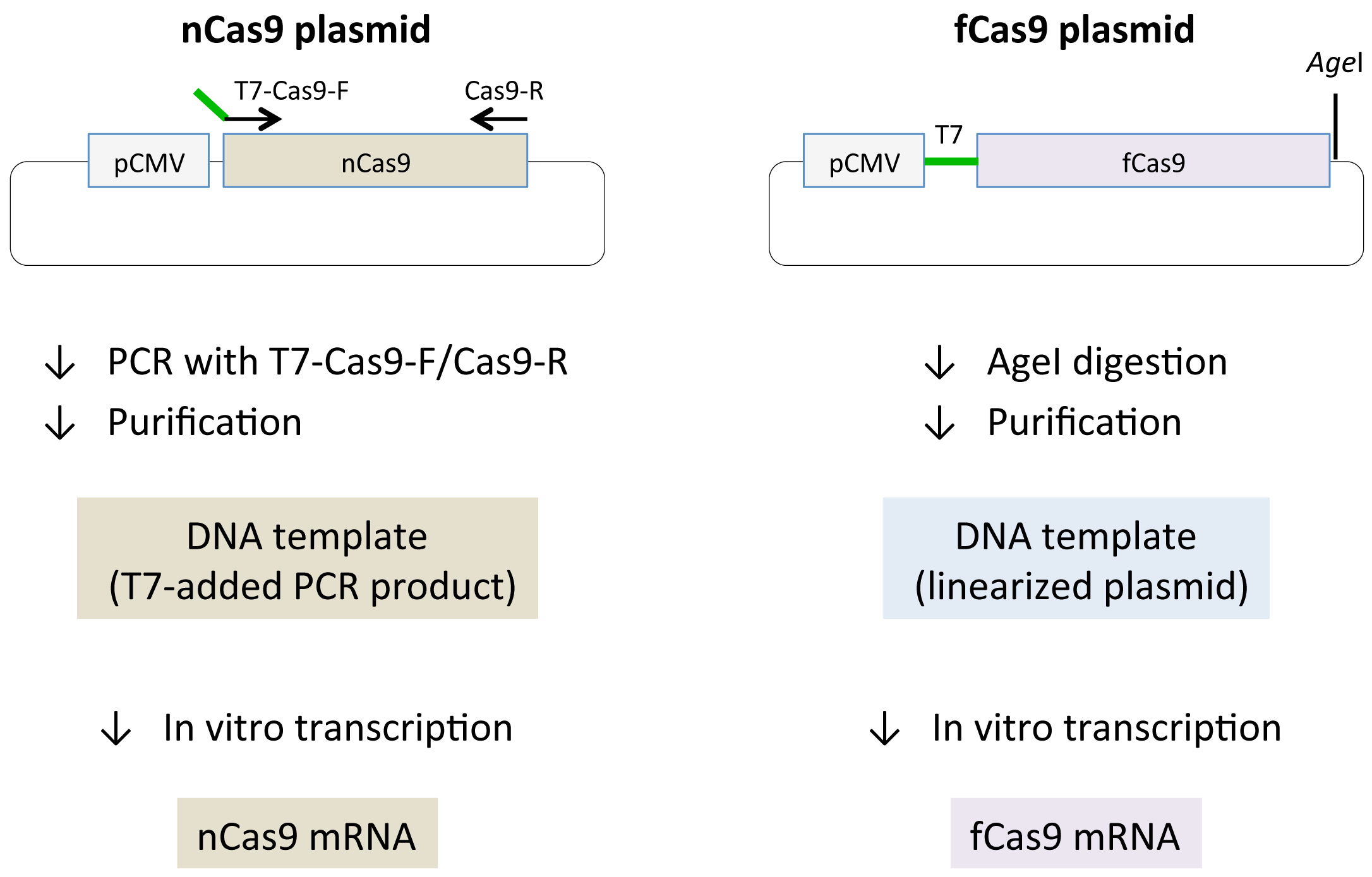
Figure 2. Preparation of Cas9 mRNA. Schematic representations of Cas9 mRNA synthesis. Black lines and arrows show backbone vector and primers respectively. Cytomegalovirus promoter (pCMV), nCas9 and fCas9 are boxed. T7 promoter sequences are depicted with green lines. (Left) Since nCas9 vector (Addgene #41816) does not contain T7 promoter, T7 promoter is added to the nCas9 coding sequence by PCR using T7-Cas9-F/Cas9-R primers and the plasmid as a template. PCR products are then subjected to in vitro transcription reaction. (Right) fCas9 vector (Addgene #52970) includes T7 promoter, which can be used for in vitro transcription reaction after linearization of the plasmid by AgeI digestion.
- Synthesize Cas9 mRNA in an in vitro transcription reaction using T7 RNA polymerase. Note, the plasmid that contains fCas9 has a T7 promoter sequence, but the plasmid that contains nCas9 does not (Figure 2). To add the T7 promoter sequence to the 5’ terminal of nCas9, amplify nCas9 by PCR with the following primers:
- In vitro transcription reactions are performed using the mMESSAGE/mMACHINE T7 Transcription Kit (according to the manufacturer’s instructions) with 2 µg of the PCR product or the linearized plasmid as template. After TURBO DNase treatment (included in the mMESSAGE/mMACHINE T7 Transcription Kit), the transcribed RNA products are purified with the MEGAclear Transcription Clean-Up Kit. We adjust the concentration of the mRNA product to 1 µg/µl.
- For microinjection, RNAs are mixed as follows:
1 µg/µl of crRNA1 and crRNA2 2.84 µl each
1 µg/µl of tracrRNA 9.32 µl
1 µg/µl of nCas9 or fCas9 mRNA 7.5 µl
RNase-free water 7.5 µl
Total volume = 30 µl
Notes:- Final concentrations of crRNA1, crRNA2, tracrRNA and Cas9 mRNA are 94.7, 94.7, 310.7 and 250 ng/µl, respectively.
- The molar ratios of crRNAs and tracrRNA are adjusted to be equal in the mixture. Molar ratios were calculated by dividing RNA concentration by RNA length. Molar ratios of crRNA1, crRNA2 and tracrRNA are 94.7 ng/µl/42 nt (= 2.25), 94.7 ng/µl/42 nt (= 2.25) and 310.7 ng/µl/69 nt (= 4.5), respectively.
- Although the RNA mixture can be stored for several months at -80 °C, we recommend that the RNA mixture be prepared fresh each time.
- Final concentrations of crRNA1, crRNA2, tracrRNA and Cas9 mRNA are 94.7, 94.7, 310.7 and 250 ng/µl, respectively.
- Preparation of DNA templates for in vitro transcription
- Collection of fertilized eggs
- On Day 1, induce superovulation in adult female mice by injecting them with 5 IU PMSG intraperitoneally. Forty-eight hours later (Day 3) inject the female mice with 5 IU hCG, then cross with adult male mice.
- On Day 4, sacrifice the female mice that were crossed on Day 3 and collect fertilized eggs from their oviducts. To remove cumulus cells, treat the zygotes with 0.3 mg/ml of hyaluronidase in M2 medium (see Recipes) at 37 °C for 1-5 min.
- Pick zygotes using mouth pipettes and then wash several times by pipetting in KSOM medium.
- On Day 1, induce superovulation in adult female mice by injecting them with 5 IU PMSG intraperitoneally. Forty-eight hours later (Day 3) inject the female mice with 5 IU hCG, then cross with adult male mice.
- Preparation of holding and injection pipettes
- Glass capillaries are drawn out using a capillary puller.
- Holding and injection pipettes are prepared from drawn-out pipettes using a microforge.
- Before injection, centrifuge the RNA mixture at 12,000 x g for 10 min. Fill the injection pipettes with 3-5 µl of the supernatant of the RNA mixture by microloader.
- Coat the outside of the RNA-filled injection pipettes with dichlorodimethylsilane.
- Glass capillaries are drawn out using a capillary puller.
- Microinjection
- Put the fertilized eggs in a drop of M2 medium and insert the injection pipette into the cytoplasm of zygote. Inject the RNA mixture (1-5 pl) into the cytoplasm. The amount of the injected solution can be controlled using FemtoJet.
- Pool the injected eggs in M2 medium. Wash the zygotes several times with KSOM medium. Incubate in a drop of KSOM drop at 37 °C until next day.
- Put the fertilized eggs in a drop of M2 medium and insert the injection pipette into the cytoplasm of zygote. Inject the RNA mixture (1-5 pl) into the cytoplasm. The amount of the injected solution can be controlled using FemtoJet.
- 2-cell embryo transfer
- Obtain pseudopregnant mice by crossing adult ICR females with vasectomized male mice on the same day of microinjection (Day 4).
- On the morning of Day 5, check ICR females for a vaginal plug. If the ICR female has a vaginal plug, then it is considered to be pseudopregnant.
- Isolate injected embryos that have reached the 2-cell stage by picking them up in a minimal volume of KSOM medium (10-18 embryos). Transfer these embryos into either oviduct an anesthetized, pseudopregnant female mouse.
- Nineteen days after transplantation, newborn mice should be obtained.
- Obtain pseudopregnant mice by crossing adult ICR females with vasectomized male mice on the same day of microinjection (Day 4).
- Genotyping the founder mice by PCR direct sequencing
- Isolate genomic DNA from a tail tip using a standard procedure (Nagy et al., 2003).
- Amplify the region of interest using primers designed around crRNA target sequences by performing PCR with the genomic DNA and primers spanning the crRNAs target sequences. Treat the PCR products with ExoSAP-IT (or Qiaquick PCR purification Kit) and sequence with either primer used for PCR.
- Isolate genomic DNA from a tail tip using a standard procedure (Nagy et al., 2003).
Data analysis
When a mutation is introduced heterozygously by crRNA/tracrRNA/Cas9, overlapped double peaks will be observed between the two target sites (see Figure 3). If a homozygous or mosaic mutation is introduced, then a single or more than two peaks will be observed, respectively. In addition to sequencing the direct PCR product, one can also clone the PCR product and sequence the plasmid. Note that there is a possibility of having a mosaic mutation even when a single peak is observed in the sequence at this step. It is recommended that the sequence at the site of mutation as well as any relevant phenotypes be analyzed in the F1 generation as well as or later generations, if possible.
Using this method, from our experience, approximately 30-40% and 30-60% of founder pups obtained after crRNA/tracrRAN/nCas9 and crRNA/tracrRNA/fCas9 injection, respectively, contained mutations at least one allele (Terao et al., 2016).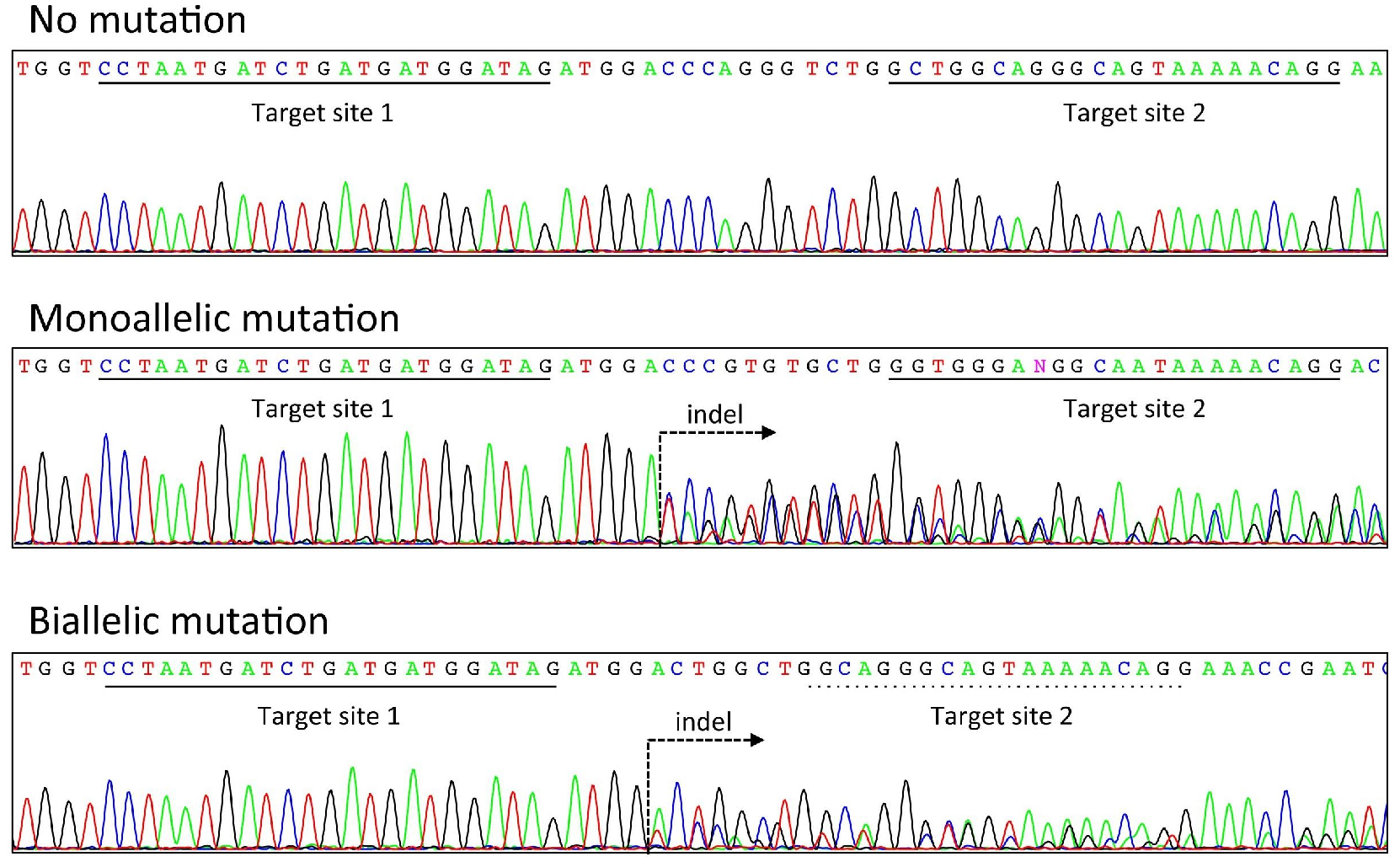
Figure 3. Example of genotyping analysis. Electropherograms of PCR products amplified from wild-type mice (top), mice carrying a mutation in a single allele (middle), and mice carrying a mutation in both alleles (bottom). Each target site of crRNA is underlined with a solid black line. If the target site is underlined with a dashed line, then it indicates that the target site is broken (see Biallelic mutation, bottom). The dashed arrow indicates a point of mutation.
Recipes
- M2 medium
94.66 mM NaCl
4.78 mM KCl
1.71 mM CaCl2
1.19 mM KH2PO4
1.19 mM MgSO4
4.15 mM NaHCO3
20.85 mM HEPES
23.28 mM sodium DL-lactate
0.33 mM sodium pyruvate
5.56 mM D-(+)-glucose
0.01% (w/v) polyvinyl alcohol
0.5% (v/v) penicillin-streptomycin
0.02% (v/v) phenol red
Adjust the pH to 7.4 with 5-10 N NaOH
Filtration with a 0.22 µm filter
Acknowledgments
This work was supported in part by a grant from the National Center for Child Health and Development, Grant Number 24-3 to S.T. This protocol is developed based on our previous work published in Exp Anim (Terao et al., 2016).
References
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A. and Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121): 819-823.
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A. and Charpentier, E. (2012). A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096): 816-821.
- Hara, S., Tamano, M., Yamashita, S., Kato, T., Saito, T., Sakuma, T., Yamamoto, T., Inui, M. and Takada, S. (2015). Generation of mutant mice via the CRISPR/Cas9 system using FokI-dCas9. Sci Rep 5: 11221.
- Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., Norville, J. E. and Church, G. M. (2013). RNA-guided human genome engineering via Cas9. Science 339(6121): 823-826.
- Nagy, A., Gertsenstein, M., Vintersten, K. and Behringer, R. (2003). Manipulating the mouse embryo: A laboratory manual, 3rd edition. Cold Spring Harbor Laboratory Press.
- Ran, F. A., Hsu, P. D., Lin, C. Y., Gootenberg, J. S., Konermann, S., Trevino, A. E., Scott, D. A., Inoue, A., Matoba, S., Zhang, Y. and Zhang, F. (2013). Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154(6): 1380-1389.
- Terao, M., Tamano, M., Hara, S., Kato, T., Kinoshita, M. and Takada, S. (2016). Utilization of the CRISPR/Cas9 system for the efficient production of mutant mice using crRNA/tracrRNA with Cas9 nickase and FokI-dCas9. Exp Anim 65(3): 275-283.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Hara, S., Terao, M. and Takada, S. (2017). A Protocol for Production of Mutant Mice Using Chemically Synthesized crRNA/tracrRNA with Cas9 Nickase and FokI-dCas9. Bio-protocol 7(11): e2340. DOI: 10.21769/BioProtoc.2340.
Category
Molecular Biology > DNA > Mutagenesis
Molecular Biology > RNA > Transfection
Cell Biology > Cell engineering > CRISPR-cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.