- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Chromatin Immunoprecipitation Experiments from Whole Drosophila Embryos or Larval Imaginal Discs

Published: Vol 7, Iss 11, Jun 5, 2017 DOI: 10.21769/BioProtoc.2327 Views: 13686
Reviewed by: Jihyun KimLeonardo G. GuilgurAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

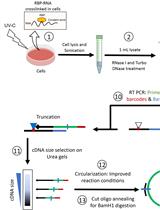
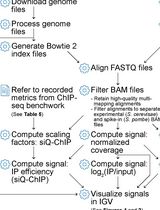
Abstract
Chromatin Immunoprecipitation coupled either to qPCR (qChIP) or high-throughput sequencing (ChIP-Seq) has been extensively used in the last decades to identify the DNA binding sites of transcription factors or the localization of various histone marks along the genome. The ChIP experiment generally includes 7 steps: collection of biological samples (A), cross-linking proteins to DNA (B), chromatin isolation and fragmentation by sonication (C), sonication test (D), immunoprecipitation with antibodies against the protein or the histone mark of interest (E), DNA recovery (E), identification of factor-associated DNA sequences by PCR or sequencing (F). The protocol described here can readily be used for ChIP-seq and ChIP-qPCR experiments. The entire procedure, describing experimental setup conditions to optimize assays in intact Drosophila tissues, can be completed within four days.
Keywords: ChIPBackground
Despite the fact that immortalized cultured cells are extensively used to study the chromatin landscape of various cell types, valuable methods for probing interaction in vivo, under physiological conditions, are necessary to perform temporal or spatial comparative analysis of transcription factor and histone modification maps between different stages of Drosophila development or between different tissues. Here we present a detailed ChIP protocol that has been optimized to work on whole Drosophila embryos and larval imaginal discs, highlighting critical experimental parameters.
Materials and Reagents
- Imaginal discs dissection step
1.Sterile tips for micropipettes, 1,000 µl
2.1.5 ml DNA low binding tubes (Eppendorf, catalog number: 022431021 )
3.Schneider’s insect medium (Sigma-Aldrich, catalog number: S0146 ) - Embryos collection step
- Petri dishes
- 1.5 ml DNA low binding (LoBind) tubes (Eppendorf, catalog number: 022431021 )
- Drosophila embryos
- SAF-INSTANT yeast (Lesaffre)
- Liquid nitrogen
- Agar
- 10% Moldex (Methyl-4-hydroxybenzoate) (Sigma-Aldrich, catalog number: W271004 ) dissolved in water
- Neutral red (Sigma-Aldrich, catalog number: N4638 )
- Egg laying medium (see Recipes)
- Petri dishes
- Chromatin immunoprecipitation protocol
- 1.5 ml DNA low binding (LoBind) tubes (Eppendorf, catalog number: 022431021 )
- 15 ml sterile plastic tubes (Greiner Bio One International, catalog number: 188261 )
- 50 ml sterile plastic tubes (Greiner Bio One International, catalog number: 227261 )
- 15 ml polystyrene Falcon tubes (Corning, catalog number: 352095 )
- Sterile filter tips, 10 µl, 200 µl, 1,000 µl (STARLAB INTERNATIONAL, catalog numbers: S1121-2710 , S1120-8810 , S1122-1830 ) (see Note 1 for more details)
- Sterile, DNase and RNase free, DNA low binding filter tips, 10 µl, 200 µl, 1,000 µl (Sorenson BioScience, catalog numbers: 35210 , 35240 , 35260 ) (see Note 1 for more details)
- Qubit® Assay tubes (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: Q32856 )
- LightCycler® 480 multiwell plate 96, white (Roche Molecular Systems, catalog number: 04729692001 )
- LightCycler® 480 sealing foil (Roche Molecular Systems, catalog number: 04729757001 )
- Liquid nitrogen
- 20% sodium dodecyl sulfate (SDS) (Biosolve, catalog number: 198123 )
- Autoclaved 1 M KCl solution
- Autoclaved 5 M NaCl solution
- Autoclaved 1 M MgCl2 solution
- Sterile filtered 1 M HEPES buffer pH 7.6 (Sigma-Aldrich, catalog number: H0887 )
- Triton X-100 (Sigma-Aldrich, catalog number: T8787 )
- 1 M DL-Dithiothreitol (DTT) prepared from powder (Sigma-Aldrich, catalog number: D0632 ) dissolved in deionized water
- 1 M sodium butyrate prepared from powder (Sigma-Aldrich, catalog number: 303410 ) dissolved in deionized water
- EDTA-free protease inhibitor cocktail tablets (Roche Molecular Systems, catalog number: 04693132001 )
- 16% formaldehyde solution, methanol-free (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 28906 )
- 2.5 M glycine prepared from powder (Sigma-Aldrich, catalog number: G8898 ) dissolved in deionized water filtered with a disposable syringe filter (0.45 μm) (Pall, catalog number: 4422 )
- 0.5 M EDTA pH 8.0 (Sigma-Aldrich, catalog number: 03690 )
- 0.5 M EGTA pH 8.0 prepared from powder (Sigma-Aldrich, catalog number: E3889 ) dissolved in deionized water
- 1 M sodium deoxycholate prepared from powder (Sigma-Aldrich, catalog number: D5670 ) dissolved in deionized water
- 20% N-Lauroylsarcosine sodium salt solution (Sigma-Aldrich, catalog number: L7414 )
- 10 mg/ml RNase A (Sigma-Aldrich, catalog number: R6513 ) dissolved in deionized water
- UltraPureTM phenol:chloroform:isoamyl alcohol (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15593049 ) referred as phenol-chloroform in the text
- 3 M sodium acetate, pH 5.5 (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9740 )
- 35 mg/ml glycogen (MP-Biomedicals, catalog number: 11GLYCO001 )
- Ethanol absolute AnalaR NORMAPUR® (VWR, catalog number: 20821.310 )
- Agarose powder (Sigma-Aldrich, catalog number: A9539 )
- 1x TAE buffer prepared from 50x solution (Biosolve, catalog number: 205023 )
- Autoclaved Tris HCl pH 8.0
- Ethidium bromide (Sigma-Aldrich, catalog number: E1510 )
- Nuclease free water, not DEPC treated (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9937 )
- Urea (MP Biomedicals, catalog number: 821519 )
- Sucrose (Sigma-Aldrich, catalog number: S0389 )
- Bromophenol blue (Sigma-Aldrich, catalog number: B5525 )
- 100 bp DNA ladder (New England Biolabs, catalog number: N3231S )
- Qubit® dsDNA HS Assay Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32854 )
- Dynabeads® Protein A for Immunoprecipitation (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10001D ) or Dynabeads® Protein G for Immunoprecipitation (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10003D ) (see Note 2 for details)
- 20 mg/ml proteinase K (Sigma-Aldrich, catalog number: P2308 )
- 1 g/µl of normal rabbit IgG (Cell Signaling Technology, catalog number: 2729 )
- Sterile filtered water (Sigma-Aldrich, catalog number: W3500 )
- LightCycler® 480 SYBR Green I Master (Roche Molecular Systems, catalog number: 04707516001 )
- 10x embryo buffer stock solution (see Recipes)
- Embryo buffer (see Recipes)
- Buffer A1 (see Recipes)
- Lysis buffer 1 (see Recipes)
- Lysis buffer 2 (see Recipes)
- Lysis buffer wash (see Recipes)
- TE buffer
- Low blue loading buffer (see Recipes)
- Elution buffer 1 (see Recipes)
- Elution buffer 2 (see Recipes)
- 1.5 ml DNA low binding (LoBind) tubes (Eppendorf, catalog number: 022431021 )
Equipment
- Sieves (pores < 0.5 mm)
- Microwave
- 1 L beaker (Corning, PYREX®, catalog number: 1003-1L )
- Magnetic stirrer
- Cages (Dutscher Scientific, catalog number: 789092 )
- Wash bottle
- Paintbrush (Carolina Biological Supply, catalog number: 173094 )
- Calibration checked pipettes 0-2 µl, 1-10 µl, 20-200 µl, 100-1,000 µl
- Rotating wheels, one in a cold room and the other one at room temperature
- Stereomicroscope
- Fine #55 forceps for dissection (Fine Science Tools, catalog number: 11255-20 )
- WheatonTM Tenbroeck tissue grinder (WHEATON, catalog number: 357422 )
- Agarose gel caster
- Shaker (For instance the Orbital Shaker from Starlab group) (STARLAB INTERNATIONAL, model: OrbitalTM Shaker, catalog number: N2400-8030 )
- Cold centrifuge (4 °C, able to reach at least 14,000 x g)
- Cold sonicator (we recommend the Bioruptor® from Diagenode, model: Bioruptor® Plus sonication device , catalog number: B01020001)
- Magnetic rack (Thermo Fisher Scientific, model: DynaMagTM-2 Magnet, catalog number: 12321D )
- Gel visualization system (UV lamp)
- Qubit® fluorometer (Thermo Fisher Scientific, InvitrogenTM, model: Qubit® 3.0 , catalog number: Q33216)
- LightCycler® Instrument II (Roche Molecular Systems, model: LightCycler® 480 Instrument II , catalog number: 05015278001)
Procedure
- Collection of biological samples
Note: This step differs for embryos and imaginal discs.
EMBRYOS:- Melt egg laying medium using a microwave (see Recipes).
- Fill the Petri dishes with 15 ml of egg laying medium.
- While egg laying medium is cooling down, fill a beaker with 100 ml of sink water and put it on a magnetic stirrer.
- Add progressively 50 g of yeast to the beaker. Achieve the mixing by hand stirring as the yeast mixture becomes very viscous.
- Once the egg laying medium gets solid, spread 5-10 ml of yeast mixture on each dish (see Figure 1).
Figure 1. Egg laying medium covered with yeast mixture - Assemble the Petri dishes onto the cages (see Figure 2) and let flies lay eggs as long as required to get a sufficient amount of embryos (see Figure 3).
Note: See Note 3 to know the number of embryos required.

Figure 2. Petri dish assembled onto the bottom of a cage containing adult flies
Figure 3. Petri dish layered with Drosophila embryos (O/N egg laying) - Collect the embryos in a sieve by spraying 1x embryo buffer with a wash bottle (see Figure 4). Use a paintbrush to help.

Figure 4. Embryos in a sieve (O/N collection) - Wash the embryos three times by putting the sieve in a bath of embryo buffer (use the cap of a tip box).
- Collect the embryos with a cut 200 µl tip and transfer them in an Eppendorf tube.
- Remove as much buffer as possible and store them at -80 °C.
IMAGINAL DISCS:
- Dissect imaginal discs in Schneider’s medium (SM) (Spratford and Kumar, 2014), and put them in a 1.5 ml LoBind Eppendorf tube, kept on ice and containing 1 ml of SM.
Note: See Note 4 to know the number of discs required. - Dissection time should not exceed 1 h.
- Centrifuge for 2 min (4 °C, 4,000 x g) and remove the supernatant. Imaginal discs can be snap frozen in liquid nitrogen and stored at -80 °C for months, allowing multiple rounds of dissection in order to collect enough material.
- Melt egg laying medium using a microwave (see Recipes).
- Cross-linking proteins to DNA
Note: At this step, embryos and imaginal discs samples are processed the same way.- If samples have been frozen, thaw them on ice for a couple of minutes.
- Resuspend the tissues and pool them in a final volume of 888 µl of ice-cold A1 buffer.
- Transfer them into a 2 ml tight Tenbroeck at room temperature (RT).
- Fixation: quickly add 112 µl of 16% formaldehyde (FA) in the Tenbroeck (final concentration of 1.8% FA). Fixation is performed at RT.
Note: See Note 5 for the concentration of FA to be used. - Disrupt the tissues for 5 min by slowly moving the pestle of the Tenbroeck up and down in the mortar.
- Transfer the disrupted tissues back to a 1.5 ml LoBind Eppendorf tube.
- Put on a rotating wheel at RT: total time for fixation is 15 min.
- Stop crosslinking: add 160 µl of 2.5 M glycine (350 mM final) and put on a rotating wheel for 5 min, then put on ice.
Note: All the following steps are done on ice or at 4 °C unless otherwise specified.
- Centrifuge for 2 min (4 °C, at 2,000 x g) and discard the supernatant.
- Wash with 1 ml of buffer A1 (disrupt the pellet by pipetting).
- Centrifuge for 2 min (4 °C, 2,000 x g).
- Repeat steps B9 and B10 twice.
- Wash once with lysis buffer 1, centrifuge for 2 min (4 °C, 2,000 x g) and remove the supernatant.
- If samples have been frozen, thaw them on ice for a couple of minutes.
- Chromatin isolation and fragmentation by sonication
Note: This step differs for embryos and imaginal discs samples.
EMBRYOS:- Prepare 2 ml of 0.1% SDS lysis buffer 2 (10 µl of 20% SDS + 1,990 µl of lysis buffer 2).
- Resuspend the pellet with (Number of embryos/1,000) x 250 µl of freshly prepared 0.1% SDS lysis buffer 2.
- Incubate for 2 h on ice, and put the ice bucket with the samples in a shaker (600 rpm) to avoid SDS precipitation.
Note: See Note 6 for an alternative method. - Sonication: split lysate in several 15 ml polystyrene Falcon tubes in order to get 1.5 ml of lysate per tube.
Note: See Note 7 for considerations about sonication efficiency. - Set the Bioruptor sonicator as follows: Number of cycles = 12, power = HIGH, ON = 30 sec, OFF = 30 sec.
- After sonication, pool all the tubes back together. At this step, samples can be snap frozen in liquid nitrogen and conserved at -80 °C for months.
IMAGINAL DISCS:- Prepare 1 ml of 1% SDS lysis buffer 2 (50 µl of 20% SDS + 950 µl of lysis buffer 2).
- Resuspend the pellet with (Number of imaginal discs/600) x 150 µl of freshly prepared 1% SDS lysis buffer 2.
- Incubate for 2 h on ice, and put the ice bucket with the samples in a shaker (600 rpm) to avoid SDS precipitation.
Note: See Note 6 for an alternative method. - Sonication: split lysate in several 1.5 ml LoBind tubes in order to get 150 µl of lysate per tube.
Note: See Note 7 for considerations about sonication efficiency. - Set the Bioruptor sonicator as follows: Number of cycles = 18, power = HIGH, ON = 30 sec, OFF = 30 sec.
- After sonication, pool all the tubes back together. At this step, samples can be snap frozen in liquid nitrogen and conserved at -80 °C for months.
- Prepare 2 ml of 0.1% SDS lysis buffer 2 (10 µl of 20% SDS + 1,990 µl of lysis buffer 2).
- Sonication test
Note: At this step, embryos and imaginal discs samples are processed the same way. If your sonication conditions are well established, you can skip this step.- For the sonication test, transfer 5-10 µl of the lysate (200 ng of DNA is sufficient) in a fresh 1.5 ml Lobind tube. During the sonication test, keep the remaining lysate at 4 °C.
- Bring the volume up to 100 µl with lysis buffer 1.
- Add 2 µl of 10 mg/ml RNase A and incubate for 6 h (to O/N) at 65 °C with 450 rpm agitation.
- Add 100 µl of phenol-chloroform and vortex for 30 sec.
- Centrifuge for 10 min (RT, maximum speed).
- Transfer the aqueous phase to a fresh 1.5 ml Lobind tube and add 10 µl of 3 M NaAc (pH 5.5), 2 µl of 35 mg/ml glycogen and 250 µl of 100% EtOH. Vortex and incubate for 30 min at -20 °C.
- Centrifuge for 20 min (4 °C, maximum speed).
- During centrifugation, prepare a 1.5% agarose gel by dissolving 0.75 g of agarose powder with 50 ml of 1x TAE buffer.
- Melt the agarose solution in a microwave, and let it cool down for 5 min before adding 3 µl of ethidium bromide.
- Cast the gel in an agarose gel caster and let it cool down at RT.
- Once the 20 min centrifugation is over, remove the supernatant and air dry the pellet for 10 min at RT.
- Resuspend the pellet in 8 µl of nuclease free H2O and add 2 µl of low blue loading buffer (see Recipes for details).
- Deposit 5 µl of 100 bp DNA ladder and the 10 µl of the sonication test on the gel.
- Run the gel for 20-25 min at 135 V and observe it using a UV lamp. An example of efficient sonication is depicted in Figure 5.
Note: See Note 8 for considerations about the importance of sonication quality.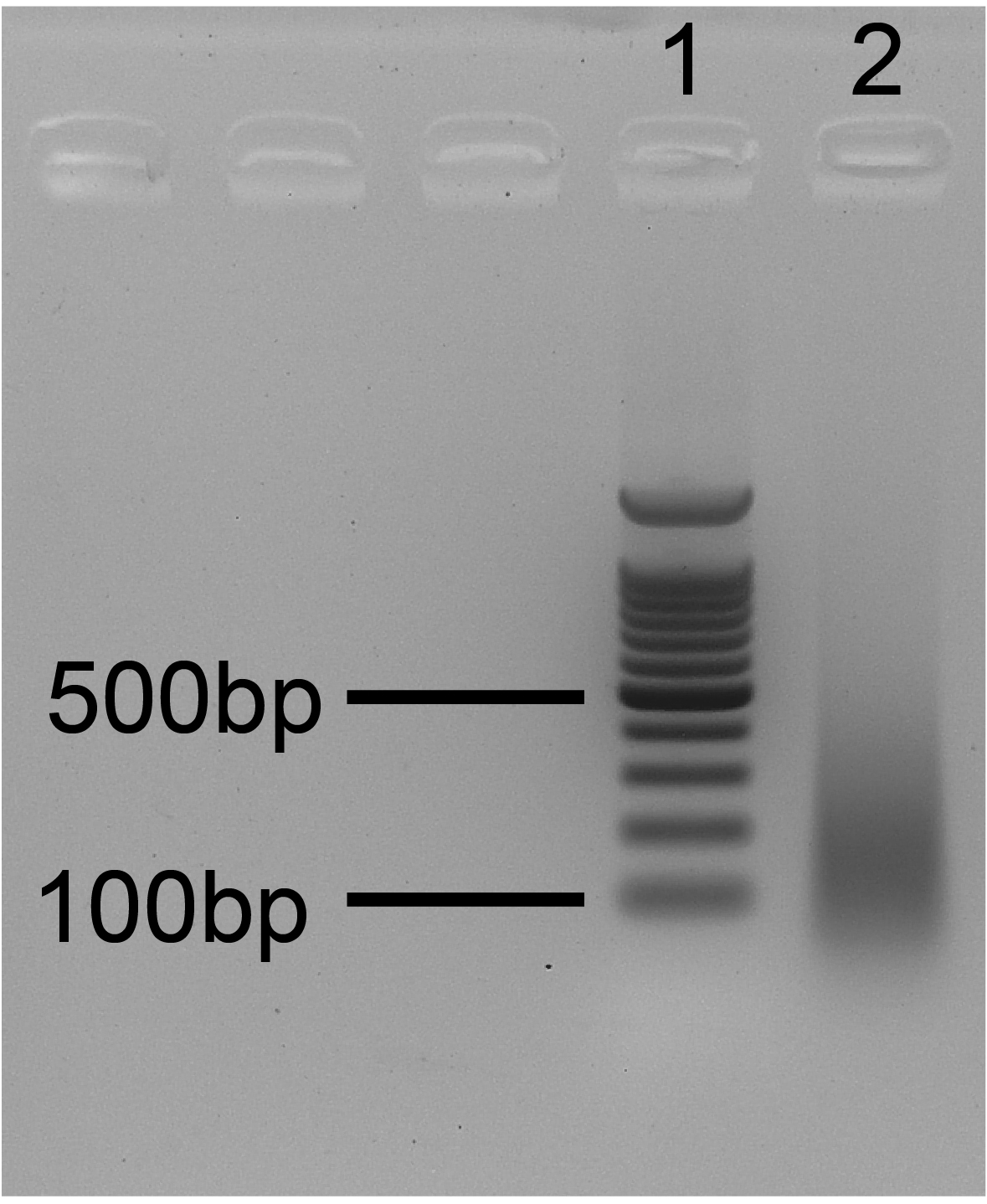
Figure 5. Ideal sonication gel. 1. 100 bp ladder; 2. Sonication test. The size of bulk DNA fragments ranges from 100 to 300 bp.
- For the sonication test, transfer 5-10 µl of the lysate (200 ng of DNA is sufficient) in a fresh 1.5 ml Lobind tube. During the sonication test, keep the remaining lysate at 4 °C.
- Immunoprecipitation
Note: This step differs for embryos and imaginal discs samples.
EMBRYOS:- If the sonication pattern is adequate, centrifuge the sonicated lysate for 5 min (4 °C, 14,000 x g). If sonication is insufficient, sonicate the chromatin for 2 cycles more with the same settings and repeat the sonication test.
- Transfer the supernatant into a fresh tube (if possible LoBind).
- Quantify the DNA using the Qubit® dsDNA HS Assay Kit, following manufacturer’s instructions (1 µl of sonicated lysate should be enough).
Note: See Note 9 for recommendations about the amount of DNA to be used for IPs. - Split chromatin samples into N aliquots, each containing 250 µl. N = number of IPs + 2 (One Mock and one INPUT).
- Wash (number of samples + 2) x 15 µl of Protein A or G Dynabeads.
Note: See Note 10 to know how to wash the beads and see Note 2 to know the type of Dynabeads to be used (A or G). - Preclearing: Add 15 µl of washed Protein A or G Dynabeads to the chromatin samples and preclear chromatin for 2 h (to O/N) at 4 °C on a rotating wheel.
- Put the tubes in the magnetic rack and transfer the precleared chromatin (supernatant) in fresh 1.5 ml LoBind tubes.
- Store the tube(s) containing the Input at 4 °C until step F13 (DNA recovery).
- Immunoprecipitation: Add the antibodies in the IP tubes and the Mock; incubate for 4 h at 4 °C on a rotating wheel. For the Mock, use the same amount of normal IgG from the same species that has been used for the IPs.
Note: See Material and Reagents for the catalog number of the rabbit normal IgG we use. See Notes for details about the quantity of antibody to use (Note 11) and which controls can be performed (Notes 12, 13 and 14). - During incubation: wash (number of IPs + 1 Mock) x 30 µl of Protein A or G Dynabeads.
Note: See Note 10 to know how to wash the beads and see Note 2 to know the type of Dynabeads to be used (A or G). - Add 30 µl of Protein A Dynabeads per sample and incubate O/N at 4 °C on a rotating wheel.
IMAGINAL DISCS:- If the sonication pattern is adequate, centrifuge the sonicated lysate for 5 min (4 °C, 14,000 x g). If sonication is insufficient, sonicate the chromatin for 5 cycles more with the same settings and repeat the sonication test.
- Transfer the supernatant into a fresh 1.5 ml LoBind tube.
- Quantify the DNA using the Qubit® dsDNA HS Assay Kit, following manufacturer’s instructions (1 µl of sonicated lysate should be enough).
Note: See Note 9 for recommendations about the amount of DNA to be used for IPs. - Split chromatin samples into N aliquots, each containing 100 µl. N = number of IPs + 2 (One Mock and one INPUT).
- Dilute 10 times by adding 900 µl of lysis buffer 2 (without SDS) in order to reach a 0.1% SDS final concentration. Final volume is 1 ml.
- Wash (number of samples + 2) x 15 µl of Protein A or G Dynabeads.
Note: See Note 10 to know how to wash the beads and see Note 2 to know the type of Dynabeads to be used (A or G). - Preclearing: Add 15 µl of washed Protein A or G Dynabeads to the chromatin samples and preclear chromatin for 2 h (to O/N) at 4 °C on a rotating wheel.
- Put the tubes in the magnetic rack and transfer the precleared chromatin (supernatant) in fresh 1.5 ml LoBind tubes.
- Store the tube(s) containing the Input at 4 °C until step F13 (DNA recovery).
- Immunoprecipitation: Add the antibodies in the IP tubes and the Mock; incubate for 4 h at 4 °C on a rotating wheel. For the Mock, use the same amount of normal IgG from the same species that has been used for the IPs.
Note: See Materials and Reagents for the catalog number of the rabbit normal IgG we use. See Notes for details about the quantity of antibody to use (Note 11) and which controls can be performed (Notes 12, 13 and 14). - During incubation: wash (number of IPs + 1 Mock) x 30 µl of Protein A or G Dynabeads.
Note: See Note 10 to know how to wash the beads and see Note 2 to know the type of Dynabeads to be used (A or G). - Add 30 µl of Protein A Dynabeads per sample and incubate O/N at 4 °C on a rotating wheel.
- If the sonication pattern is adequate, centrifuge the sonicated lysate for 5 min (4 °C, 14,000 x g). If sonication is insufficient, sonicate the chromatin for 2 cycles more with the same settings and repeat the sonication test.
- DNA recovery
Note: At this step, embryos and imaginal discs samples are processed in the same way.- The next day, put the tubes on the magnetic rack and discard the supernatant.
- Put 1 ml of lysis buffer wash (kept on ice) in the tubes containing the IPs and the Mock, remove them from the magnetic rack and put them back on ice.
- Homogenize by pipetting and incubate for 5 min at 4 °C on a rotating wheel.
- Put the tubes on a magnetic rack and remove the supernatant.
- Repeat steps F2-F4 three times with lysis buffer wash (4 washes in total) and two times with TE buffer (kept on ice).
- After the last wash, be careful to remove all the remaining TE buffer.
- First elution: Add 100 µl of elution buffer 1 and homogenize by pipetting.
- Incubate the IPs and the Mock at 65 °C for 15 min under vigorous shaking (1,300 rpm).
- Put the samples on the magnetic rack and transfer the supernatants in fresh 1.5 ml LoBind tubes.
- Second elution: Add 150 µl of elution buffer 2 to the beads and homogenize by pipetting.
- Incubate at 65 °C for 15 min under vigorous shaking (1,300 rpm).
- Put the tubes on a magnetic rack and pool the two eluates (final volume 250 µl).
- Decrosslinking: incubate all the samples for 6 h (to O/N) at 65 °C, 300 rpm. Don’t forget to also take the Input(s) that was/were kept at 4 °C.
- The day after, add 3 µl of 20 mg/ml Proteinase K per tube and incubate for 2 h at 56 °C.
- Add 1 volume of phenol-chloroform and vortex for 30 sec.
- Centrifuge for 15 min (RT, maximum speed).
- Transfer aqueous phase to fresh 1.5 ml LoBind tubes and add 1/10 volume of 3 M NaAc, pH 5.5 (25 µl), 2 µl of 35 mg/ml glycogen and 2.5 volumes of 100% EtOH (625 µl).
- Precipitation: vortex the samples and incubate them O/N at -20 °C.
- The next day, put the tubes on the magnetic rack and discard the supernatant.
- Identification of factor associated DNA sequences by PCR or sequencing
Note: At this step, embryos and imaginal discs samples are processed in the same way.- The day after, centrifuge for 30 min at full speed at 4 °C.
- Discard supernatant and wash the pellet with 500 µl of ice-cold 75% EtOH.
- Centrifuge for 10 min (4 °C, maximum speed).
- Discard supernatant and air dry the pellet for 10 min.
- Resuspend the pellet in the required volume of nuclease free H2O (depending on the downstream applications, see Note 15 for more details) and put the tubes at 37 °C for 10 min, 450 rpm.
- For ChIP-Seq, see Note 16.
- For qPCR, we use 5 µl of Master Mix, 0.1 µl of each primer (50 pmol/µl), 2.8 µl of water and 2 µl of DNA sample.
- Before deposition on the PCR plate, the Input is diluted 10 times to keep the same range of Crossing point (Cp) with the one of the IPs.
- See Data analysis section to know how to analyze the results.
- The day after, centrifuge for 30 min at full speed at 4 °C.
Data analysis
- It is very important to carefully select the target regions of the oligo pairs. We recommend to use at least two negative controls: one locus that is known to not be enriched for the studied mark/protein, and another one located about 1-5 kb upstream or downstream of the region of interest. A positive control region is also required. Ideally, this region is known to be enriched for the studied mark/protein.
- Perform at least two technical duplicates for each oligo pairs.
- Once Crossing points (Cps) have been acquired, calculate the mean between the two technical replicates (see Figure 6A):
MEAN = 2^-(mean(Cp1,Cp2)) to go back to the linear scale. - For each target region, calculate the % of Input for the IP and the MOCK (see Figure 6A):
% of Input IP= 2^-(MEAN IP)/2^-(MEAN INPUT/10)
% of Input MOCK= 2^-(MEAN MOCK)/2^-(MEAN INPUT/10)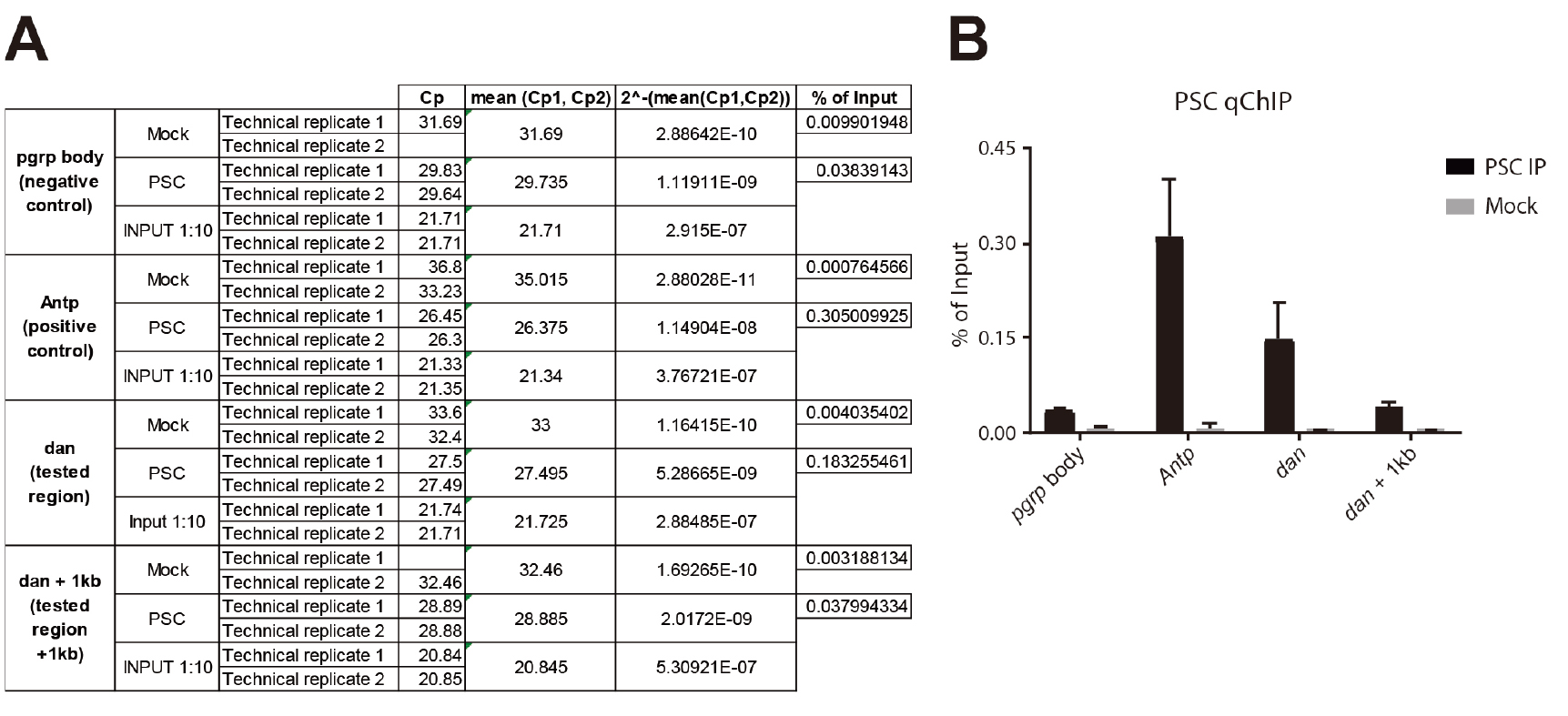
Figure 6. PSC ChlP from Eye-Antennal imaginal discs. A. Excel sheet showing the different calculation steps (columns) for replicate 1; B. Histogram showing the relative enrichment of each region expressed in % of Input. The different target regions are represented on the x-axis, and the y-axis corresponds to the % of Input. ‘pgrp body’ is used as a negative control region and is located in the body of the PGRP-LE gene. Antp (Antennapedia) is a well characterized PSC target. dan (tested region) is significantly enriched compared to the negative control. dan is also significantly enriched compared to a region located 1 kb downstream (dan + 1 kb), showing that regions sharing the same higher-order chromatin environment can have different local composition, and providing a further and ideal control. - Results can finally be displayed on a bar plot (see Figure 6B).
- Figure 6B shows a qChIP experiment that was performed in triplicate from eye-antennal imaginal discs (500 discs per IP). The PSC antibody was described in (Strutt and Paro, 1997) and 10 µl was used per IP. The following PCR primers were used for qPCR:
pgrp body: →F:cctggtgaatgatagcttactctg
→R:cttactcaaaaccgaagagatcg
Antp: →F:tggccgagtttatatcgaagcg
→R:cggccaacttgtgttgttgttc
dan: →F:cattgtacatttggctgtcacc
→R:gtcagttggtctgccttggtat
dan + 1 kb: →F:ctctctagcttgacgcaccttt
→R:aaactgtttagacccgacatgg - The Mock corresponds to the background level and should therefore be consistently lower than the IP. The enriched regions are the ones that show an enrichment compared to the negative control regions, with no enrichment in the Mock.
Notes
- Sterile, DNase and RNase free, DNA low binding filter tips have to be used when the tip gets in contact with the solubilized chromatin. Otherwise, use sterile filter tips.
- Protein A Dynabeads are more suitable to work with rabbit IgG while Protein G is preferentially used for mouse IgG.
- For 16-18 h old embryos, use at least 500 adult flies and let them lay eggs for 2 h. Remove the adults from the cage and let the embryos develop for 16 h. 1,000 embryos (roughly 50 mg wet weight) are sufficient per IP (histone marks and/or proteins).
- For proteins, 400 imaginal discs are required per IP (5 µg of DNA after lysis). For histone marks, 200 are sufficient.
- This protocol works with 1.8% FA fixation for proteins. Whereas, for histone marks 1% FA is usually sufficient.
- As an alternative to the use of a shaker for the lysis step, it is possible to manually pipette the lysate every 20/30 min to avoid SDS precipitation.
- For sonication, 4 parameters are essential: 1) the temperature (the bath has to be maintained at 4 °C), 2) the % of FA used for fixation (the higher the concentration, the longer the sonication time), 3) the volume in which sonication is performed (with the Diagenode Bioruptor, we recommend to not sonicate more than 150 µl of lysate in Eppendorf tubes [imaginal discs] and 1.5 ml in 15 ml polystyrene Falcon [embryos]), 4) the concentration of SDS in the lysis buffer (more SDS means easier sonication).
- The quality of sonication is crucial, since it will determine the resolution of the ChIP experiment. A good average size is 200-300 bp.
- Use 2-5 µg of DNA for histone marks and 5-10 µg for proteins. Nevertheless, the quality of the IP also depends highly on the quality and the quantity of antibody used.
- Dynabeads washes:
- Before taking the beads, resuspend them carefully.
- Put the tube containing the beads on a magnetic rack and remove the buffer.
- Add 3 times the volume of lysis buffer 2 (as an example, for 75 µl of beads wash with 3 x 75 = 225 µl of lysis buffer 2), take the tubes out of the rack and homogenize by pipetting to rinse the beads.
- Put the tubes containing the beads on a magnetic rack, discard the supernatant and repeat the previous step.
- Put the tubes containing the beads on a magnetic rack and discard the buffer. Resuspend the beads in the initial volume ([number of samples + 2] x 15 µl or [number of samples + 1] x 30).
- We recommend to use 5 µg of antibody per IP, and the same quantity for the Mock.
- Mock (non-specific IgG) and Input are good controls to assess the specificity and the enrichment of the protein over the background. For ChIP-Seq, only the Input is mandatory.
- Another possible control consists in using a specific antibody for a protein/histone mark that should not colocalize with your protein/histone mark of interest.
- Ideally, the specificity of the antibody should be assessed by performing a ChIP in a K.O or K.D tissue.
- For ChIP-Seq, resuspend in 25 µl of nuclease free water. For qChIP, resuspend in the required volume to perform your measurements in duplicates.
- For ChIP-Seq, use 1 µl of DNA sample to quantify the DNA using Qubit® dsDNA HS Assay Kit. Then, use 2 µl of DNA sample dissolved in as much nuclease free water as necessary to test 3-4 loci by qPCR (don’t forget to take the dilution in account when analyzing the data). If qPCR controls are satisfying, samples can be used for library preparation and sequencing.
Recipes
- Egg laying medium (325 ml)
8.75 g of agar mixed in 275 ml of water are dissolved in a microwave. Then 48 ml of vinegar and 3.2 ml of 10% Moldex are added with 50 mg of neutral red - 10x embryo buffer stock solution
7% NaCl
0.5% Triton X-100Note: The solutions described below should not be stored for more than 1 week at 4 °C.
- Embryo buffer
Dilute 10 times the 10x stock solution with deionized water - Buffer A1
60 mM KCl
15 mM NaCl
4 mM MgCl2
15 mM HEPES
0.5% Triton X-100
0.5 mM DTT
10 mM sodium butyrate in sterile filtered water
Add one tablet of protease inhibitor cocktail each 50 ml - Lysis buffer 1
140 mM NaCl
15 mM HEPES
1 mM EDTA
0.5 mM EGTA
1% Triton X-100
0.5 mM DTT
0.1% sodium deoxycholate
10 mM sodium butyrate in sterile filtered water (Sigma-Aldrich)
Add one tablet of protease inhibitor cocktail each 50 ml
Note: Lysis buffer 1 will also be used to prepare lysis buffer 2 and lysis buffer wash. - Lysis buffer 2
0.5% N-lauroylsarcosine in FRESH lysis buffer 1 - Lysis buffer wash
0.05% SDS in FRESH lysis buffer 1 - TE buffer
0.1 mM EDTA
10 mM Tris HCl pH 8.0 in sterile filtered water (Sigma-Aldrich) - Low blue loading buffer
4 M urea
50% sucrose
1 mM EDTA
Few grains of bromophenol blue
Note: The bromophenol blue migrates at the same size than sonicated chromatin, and should therefore be kept as much diluted as possible. - Elution buffer 1
10 mM EDTA
50 mM Tris HCl pH 8.0
1% SDS in sterile filtered water - Elution buffer 2
0.1 mM EDTA
10 mM Tris HCl pH 8.0
0.67% SDS
Acknowledgments
We thank all the authors of the article from which this protocol was adapted from (Loubiere et al., 2016). We thank Frédéric Bienvenu (Institute of Functional Genomics, Montpellier, France) for his technical support. We thank Filippo Ciabrelli for discussions and help in writing this manuscript.
References
- Loubiere, V., Delest, A., Thomas, A., Bonev, B., Schuettengruber, B., Sati, S., Martinez, A. M. and Cavalli, G. (2016). Coordinate redeployment of PRC1 proteins suppresses tumor formation during Drosophila development. Nat Genet 48(11): 1436-1442.
- Spratford, C. M. and Kumar, J. P. (2014). Dissection and Immunostaining of Imaginal Discs from Drosophila melanogaster. J Vis Exp (91): 51792.
- Strutt, H. and Paro, R. (1997). The polycomb group protein complex of Drosophila melanogaster has different compositions at different target genes. Mol Cell Biol 17(12): 6773-6783.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Loubiere, V., Delest, A., Schuettengruber, B., Martinez, A. and Cavalli, G. (2017). Chromatin Immunoprecipitation Experiments from Whole Drosophila Embryos or Larval Imaginal Discs. Bio-protocol 7(11): e2327. DOI: 10.21769/BioProtoc.2327.
Category
Molecular Biology > DNA > DNA-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.