- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In utero Electroporation of Mouse Embryo Brains
Published: Vol 2, Iss 14, Jul 20, 2012 DOI: 10.21769/BioProtoc.231 Views: 29127
Original research article
The authors used this protocol in:
Jan 2010
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
This is a non-invasive technique to introduce transgenes into developing brains. In this technique, DNA is injected into the lateral ventricle of the embryonic brains, and is incorporated into the cells through electroporation. Embryos then continue their development in normal conditions in vivo. The effects of genes of interest can be evaluated at certain time points after in utero electroporation. This technique allows acute knockdown or over expression of genes of interest. Compensatory effects from other genes are less likely to happen; it also circumvents possible chronic detrimental effects.
Keywords: In utero electroporationMaterials and Reagents
- EndoFree Plasmid Kit (QIAGEN, catalog number: 12362 )
- Fast green FCF (Sigma-Aldrich, catalog number: F7252 )
- Sterile saline (0.9% sodium chloride)
- 70% ethanol
- Katemine + Xylzaine mixture (see Recipes)
Equipment
- Micropipettes (Borosilicate with filament O.D.: 1mm, I.D.: 0.78 mm, 10 cm length) (Sutter Instruments, catalog number: BF100-78-10 )
- Micropipette puller P-97/ IVF (Sutter Instruments, Novato, CA)
- Electroporator (Electro-Square porator CUY21) (NEPA Gene)
- Platinum plate tweezers-type electrode (Protech International, model: CUY650-P5 )
- Ring forceps (Fine Science Tools, catalog number: 11101-09 )
- Serrated forceps (Fine Science Tools, catalog number: 11000-12 )
- Fine scissors (Fine Science Tools, catalog number: 14060-09 )
- Needle holder (Fine Science Tools, catalog number: 12003-15 )
- Silk Black Braided Suture (Ethicon, catalog number: K871 )
- 3”x3” Sterile Gauze (Dynarex, catalog number: 3353 )
- Square pulse electroporator CUY21 (Nepagene, Japan). A foot pedal is required, because during the surgery both of your hands are occupied to hold the animal and the electrodes, respectively.
- Mouth pipet (Sigma-Aldrich, catalog number: A5177 )
- Fiber optic light Nikon MKII or any other brand)
- Vaporizer for isoflurane anesthetic (Porter Instruments Company, model: 100-F )
Isoflurane is highly recommended as anesthetics. If this is not available, intraperitoneal injection of the mixture of ketamine (80-100 mg/kg) and xylazine (5-10 mg/kg) will also work. But avoid avertin that is toxic to embryos.
Procedure
- Preparation of micropipettes for DNA injection
- Pull the borosilicated micropeppets with the Micropipette puller. The following parameters are recommended: pressure, 500; heat, 800; pull, 30; velocity, 40; time, This program should produce a pipette with long shoulder that tapers gradually.
- Cut off pulled pipettes with forceps at ~1.2 cm from the shoulder of the pipettes.
- Mark tips of cut pipettes with a water-resistant magic marker in order to clarify their ends. Mark bodies of the pipettes every 5 mm length water-resistant magic marker (1 span with 5 mm corresponds to 5 μl).
- Pull the borosilicated micropeppets with the Micropipette puller. The following parameters are recommended: pressure, 500; heat, 800; pull, 30; velocity, 40; time, This program should produce a pipette with long shoulder that tapers gradually.
- DNA preparation
- Purify plasmids using the EndoFree Plasmid Kit. The final concentration of the DNA should be higher than 1 μg μl-1. Higher concentrations of DNA produce brighter fluorescence.
- Add 1/10 volume of 1% fast green to DNA solution as a tracer (final 0.1%).
- Purify plasmids using the EndoFree Plasmid Kit. The final concentration of the DNA should be higher than 1 μg μl-1. Higher concentrations of DNA produce brighter fluorescence.
- Electroporation
- Set up the electroporator: For E15 embryos: 35 V, 50 mSec On, 950 mSec Off, 5 pulses. For younger or older animals, the voltage should decrease or increase accordingly. I usually use 1-2 V increment for each day.
- Set up the mouth pipette: Inset one pulled micropipette into the mouth pipette. Draw about 10 μl DNA solutions into the micropipette.
- Anesthetize a timed-pregnant mouse with isoflurane (or with i. p. injection of katemine + Xylazine).
- After initial anesthetization with isoflurane, put the mouse on the operation platform with the abdomen upside. Then fit the mask from the isoflurane vaporizer on the mouse nose. Tape the leg with lab tapes to the operation platform.
- Wash the abdomen with 70% ethanol. Shave the fur over the abdomen using a small razor (can be bought from Walgreen). Cover the abdomen with a piece of folded gauze which has a 3 cm long slit in its center (Figure 1).
- Damp the gauzer with sterile saline.
- Using the fine scissors cut the skin longitudinally for about 2 cm long at the midline. Then make an incision of about 1.5 cm in the muscle of the abdominal cavity. The midline (can be seen as a thin white line) of the abdominal wall should not be cut for well healing.
- With ring forceps, take out the uterus carefully by pinching gaps between embryos (but not either the placenta or embryos). It is important to take care not to damage either the placenta or the blood vessels connected with the uterus. Throughout surgery you should keep the uterus wet by applying prewarmed (37 °C) saline.
- Hold the utero gently with serrated forceps, and carefully push one embryo with the ring forceps to the uterine wall. By the illumination of the fiber optics, the uterin well is transparent. The telencephalon is clearly visible.
- Hold the embryo with the ring-forceps in one hand, and use the other hand to hold the mouth pipette. Penetrating the neocortex 2-3 mm with the micropipette, and the micropipette tip will be in the lateral ventricle (Figure 2).
- Inject 1-3 μl of the DNA solution into the ventricle. If correctly targeted, the lateral ventricle should be filled with DNA-fast green, and exhibits a crescent shape.
- Hold the DNA-injected embryo in parallel along its antero-posterior axis through the uterus with the ring forceps, and put the platinum plate tweezers-type electrode across the brain, with the “+” electrode next to the injected side. Deliver the electric pulses to the embryo: 35 V, 50 mSec On, 950 mSec Off, 5 pulses.
- Continue with another embryo. You can inject all the embryos in the litter except the one that is closest to the vagina. Avoiding that one will help to avoid abortion.
- Carefully put the embryos back to the abdominal cavity.
- Fill the cavity with warm saline. It is important to try your best to place the embryos into their original position. Filling the abdomen with PBS may help the embryos to slide back to their original position.
- Close the surgical incision in the uterine wall with suture, then suture the skin.
- Keep the animal warm at 37 °C until the recovery from anesthesia.
- Set up the electroporator: For E15 embryos: 35 V, 50 mSec On, 950 mSec Off, 5 pulses. For younger or older animals, the voltage should decrease or increase accordingly. I usually use 1-2 V increment for each day.
Representative data

Figure 1. Adapted from Saito and Nakatsuji, 2001, Dev Biol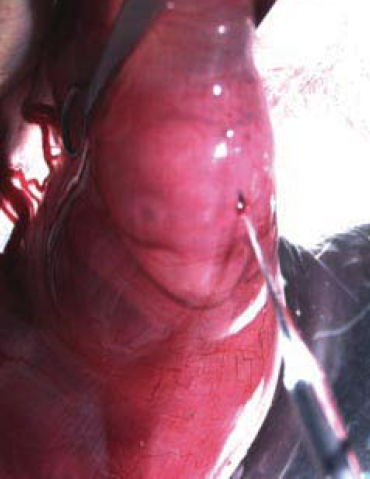
Figure 2.Adapted from Saito and Nakatsuji, 2001, Dev Biol
Recipes
- 1% Fast green in H2O (filtered and store at RT)
- Katemine + Xylzaine mixture should be made right before use:
Combine: 1 ml Ketamine (concentration 100 mg/ml)
0.5 ml Xylazine (concentration 20 mg/ml)
8.5 ml sterile saline or PBS
Dosage: 0.1 ml per 10 mg of body weight.
Acknowledgments
This protocol was adapted from Saito and Nakatsuji (2001), Ge et al. (2009) and Mao et al. (2010).
References
- Ge, X., Frank, C. L., Calderon de Anda, F. and Tsai, L. H. (2010). Hook3 interacts with PCM1 to regulate pericentriolar material assembly and the timing of neurogenesis. Neuron 65(2): 191-203.
- Mao, Y., Ge, X., Frank, C. L., Madison, J. M., Koehler, A. N., Doud, M. K., Tassa, C., Berry, E. M., Soda, T., Singh, K. K., Biechele, T., Petryshen, T. L., Moon, R. T., Haggarty, S. J. and Tsai, L. H. (2009). Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 136(6): 1017-1031.
- Saito, T. and Nakatsuji, N. (2001). Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev Biol 240(1): 237-246.
Article Information
Copyright
© 2012 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Ge, X. (2012). In utero Electroporation of Mouse Embryo Brains. Bio-protocol 2(14): e231. DOI: 10.21769/BioProtoc.231.
Category
Neuroscience > Development > Morphogenesis
Molecular Biology > DNA > Transformation
Cell Biology > Tissue analysis > Tissue isolation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.