- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Nucleosome Positioning Assay
Published: Vol 7, Iss 10, May 20, 2017 DOI: 10.21769/BioProtoc.2285 Views: 12470
Reviewed by: Antoine de MorreeXiaoyi ZhengToshitsugu Fujita
Original research article
The authors used this protocol in:

Mar 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The basic unit of chromatin is the nucleosome, a histone octamer with 147 base pairs of DNA wrapped around it. Positions of nucleosomes relative to each other and to DNA elements have a strong impact on chromatin structure and gene activity and are tightly regulated at multiple levels, i.e., DNA sequence, transcription factor binding, histone modifications and variants, and chromatin remodeling enzymes (Bell et al., 2011; Hughes and Rando, 2014). Nucleosome positions in cells or isolated nuclei can be detected by partial nuclease digestion of native or cross-linked chromatin followed by ligation-mediated polymerase chain reaction (LM-PCR) (McPherson et al., 1993; Soutoglou and Talianidis, 2002). This protocol describes a nucleosome positioning assay using Micrococcal Nuclease (MNase) digestion of formaldehyde-fixed chromatin followed by LM-PCR. We exemplify the nucleosome positioning assay for the promoter of genes encoding ribosomal RNA (rRNA genes or rDNA) in mice, which has two mutually exclusive configurations. The rDNA promoter harbors either an upstream nucleosome (NucU) covering nucleotides -157 to -2 relative to the transcription start site, or a downstream nucleosome (NucD) at position -132 to +22 (Li et al., 2006; Xie et al., 2012). Radioactive labeling of LM-PCR products followed by denaturing urea-polyacrylamide gel electrophoresis allows resolution and relative quantification of both configurations. As depicted in the diagram in Figure 1, the nucleosome positioning assay is a versatile low to medium throughput method to map discrete nucleosome positions with high precision in a semi-quantitative manner.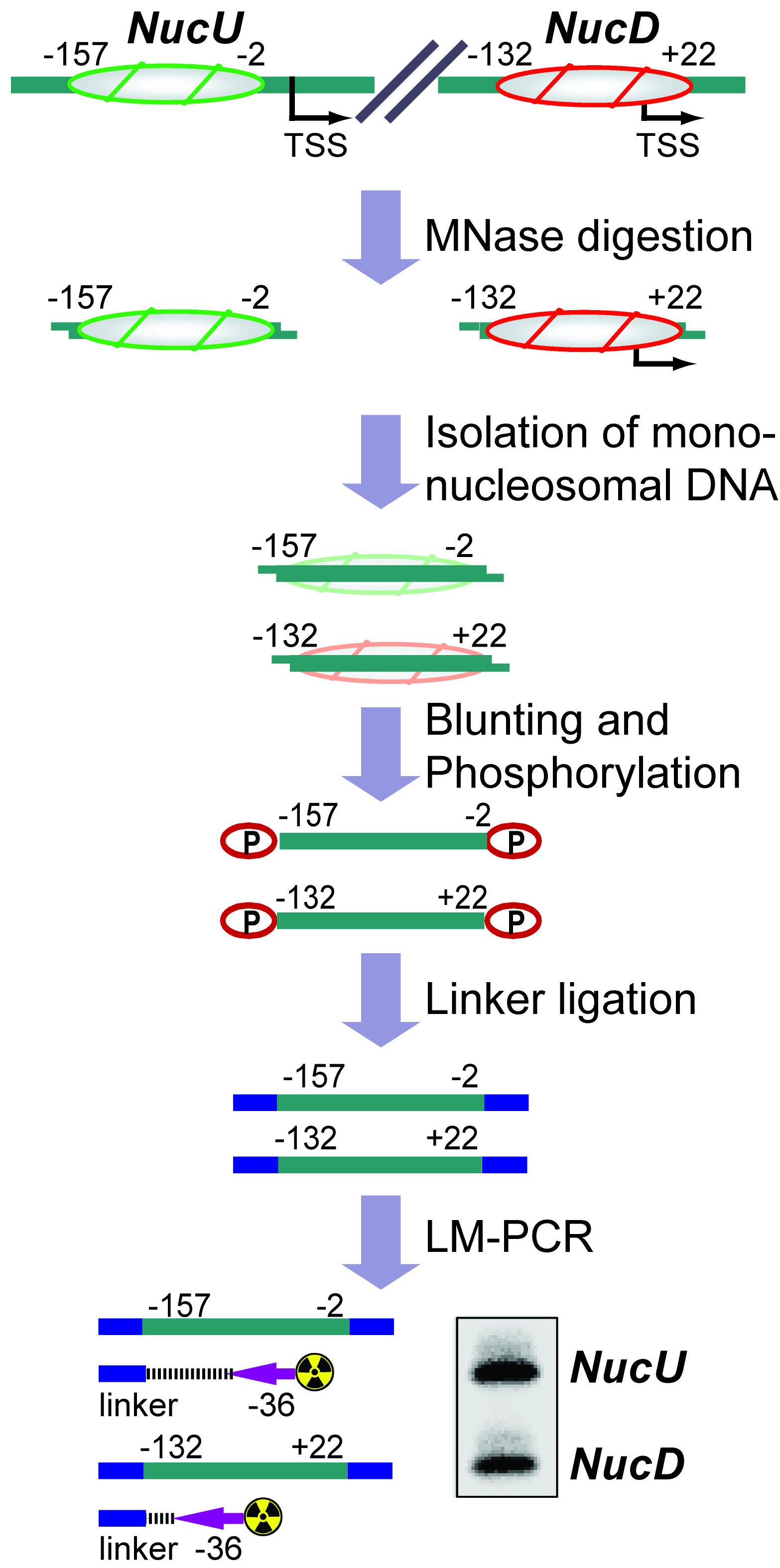
Figure 1. Flow chart depicting the nucleosome positioning assay. The diagram shows how the assay is used to detect the ratio between upstream (NucU) and downstream (NucD) nucleosome positions at the mouse rDNA promoter. After all steps have been performed, the LM-PCR yields two radiolabeled products that differ in size and correspond to NucU and NucD. Signal intensities of the bands reflect the relative abundance of each nucleosome position in the original sample.
Background
Chromatin accessibility is regulated by nucleosome packaging, which therefore directs DNA-templated reactions such as transcription, DNA repair, recombination and replication. Dynamic positioning of nucleosomes depends on DNA sequence, transcription factor binding, histone modifications, histone variants and chromatin remodeling enzymes, and is used by cells to regulate genome activity (Bell et al., 2011; Hughes and Rando, 2014). The inaccessibility of nucleosomal DNA facilitates probing of nucleosome positions in cells by digesting nucleosome-free chromatin regions with nucleases like DNase I and MNase. These, and similar approaches are nowadays frequently combined with deep sequencing methods and provide thereby a genome-wide picture of nucleosome positioning (Tsompana and Buck, 2014). However, DNase-seq and MNase-seq assays are relative labor- and cost-intensive and might be immoderate for analysis of the nucleosomal architecture of a specific genomic region. In such a case, MNase digestion of chromatin followed by LM-PCR provides a simple and straightforward alternative, which allows interrogation of nucleosome positions at a given genomic site. Here we describe this gene-centric nucleosome positioning assay and demonstrate its application for analysis of the two nucleosome configurations at the mouse rRNA gene promoter (Li et al., 2006; Xie et al., 2012; Zhao et al., 2016a and 2016b).
Materials and Reagents
- Pipette tips (TipOne filter tips, STARLAB INTERNATIONAL)
- 10 cm-dish
- Tubes (Eppendorf Safe-lock microcentrifuge tubes) (Eppendorf, catalog number: 0030120086 )
- Cell scraper (Sigma-Aldrich, catalog number: SIAL0010 )
- Syringe
- Whatman paper (Whatman, catalog number: 10547922 )
- Plastic wrap
- Clean razor blades
- Immortalized mouse embryonic fibroblast cell line NIH/3T3 (ATCC, catalog number: CRL-1658 )
- Formaldehyde solution (Sigma-Aldrich, catalog number: F8775 )
- Glycine (Sigma-Aldrich, catalog number: G7126 )
- Phosphate buffered saline (PBS)
- 0.2 M ethylene glycol-bis(2-aminoethylether)-N,N,N’,N’-tetraacetic acid (EGTA), adjust the pH to 8.0 with NaOH (Sigma-Aldrich, catalog number: E3889 )
- 0.5 M ethylenediaminetetraacetic acid (EDTA), adjust the pH to 8.0 with NaOH (Sigma-Aldrich, catalog number: E5134 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S7653 )
- Phenol:chloroform:isoamyl alcohol (25:24:1, v/v) (Carl Roth, catalog number: A156.1 )
- Sodium acetate (pH 5.2)
- 70% ethanol
- QIAquick Gel Extraction Kit (QIAGEN, catalog number: 28706 )
- Quick Blunting Kit (New England Biolabs, catalog number: E1210L )
- QIAquick PCR Purification Kit (QIAGEN, catalog number: 28106 )
- T4 DNA ligase (New England Biolabs, catalog number: M0202S )
- T4 polynucleotide kinase (New England Biolabs, catalog number: M0201S )
- PCR-primer specific for the nucleosomal region of interest. For the mouse rDNA promoter we used mrDNA (-63/-36): GATCACAAGCATAAAAGAGACAGGGAGG
- QIAquick Nucleotide Removal Kit (QIAGEN, catalog number: 28306 )
- [γ-32P]-ATP (3,000 Ci/mmol, 10 mCi/ml) (PerkinElmer, catalog number: BLU002001MC )
- GoTaq G2 Hot-Start Green PCR Master Mix (Promega, catalog number: M742A )
- Linker primers: linker S: 5’-gaattcagatc-3’, linker L: 5’-gcggtgacccgggagatctgaattc-3’
- DMSO (Sigma-Aldrich, catalog number: D8418 )
- Sucrose (Sigma-Aldrich, catalog number: 84097 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P9541 )
- 1 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) adjust the pH to 7.9 with NaOH (Applichem, catalog number: A1069 )
- Potassium phosphate dibasic (K2HPO4·3H2O) (Carl Roth, catalog number: 6878.1 )
- Magnesium chloride (MgCl2·6H2O) (Applichem, catalog number: 131396.1211 )
- Calcium chloride (CaCl2) (Sigma-Aldrich, catalog number: 499609 )
- L-α-lysophosphatidylcholine (Sigma-Aldrich, catalog number: L4129 )
- Micrococcal Nuclease (MNase) (New England Biolabs, catalog number: M0247S )
- Tris base
- Boric acid
- Formamide
- Xylene cyanol
- Bromophenol blue
- SDS (Sigma-Aldrich, catalog number: 74255 )
- Rotiphorese sequencing gel concentrate (Carl Roth, catalog number: 3043.1 )
- Rotiphorese sequencing gel diluents (Carl Roth, catalog number: 3047.1 )
- Ammonium persulfate (APS) (Carl Roth, catalog number: 9592.2 )
- TEMED (Carl Roth, catalog number: 2367.3 )
- Permeabilization buffer (see Recipes)
- MNase digestion buffer (see Recipes)
- TE buffer (see Recipes)
- 10x TBE buffer (see Recipes)
- Formamide loading buffer (see Recipes)
- Denaturing polyacrylamide gel (6%) (see Recipes)
Equipment
- Pipette
- Standard microcentrifuge
- NanoDrop 2000 UV-Vis spectrophotometer (Thermo Fisher ScientificTM, model: NanoDrop 2000 )
- PCR machine
- Electrophoresis equipment with power supply
- Vacuum pump
- Gel dryer (Bio-Rad Laboratories, model: 583 )
Note: This product has been discontinued. - Phosphor imaging instrument (FujiFilm, model: FLA-3000 )
- Imaging plate (GE Healthcare, model: BAS-IP MS 2025 E )
Software
- Quantification software (Raytest, AIDA Image Analyzer)
Procedure
- Purification of mononucleosomal DNA from cells
- Fix NIH/3T3 cells on a 10 cm-dish (80% confluency) by adding 270 μl of 37% formaldehyde solution to 10 ml culture medium (1% formaldehyde final) for 10 min at room temperature. Quench excessive formaldehyde by adding 1/20 volume of 2.5 M glycine solution (125 mM glycine final) for 5 min.
- After two washes with PBS, permeabilize cells at 37 °C for 1 min in permeabilization buffer on the culture dish.
- Remove the permeabilization buffer and directly incubate cells in MNase digestion buffer for 20 min at room temperature. Inactivate MNase by adding EDTA and EGTA to a final concentration of 10 mM each. Discard the solution and harvest cells in PBS by scraping and transfer them to a microcentrifuge tube.
- To revert formaldehyde-mediated crosslinking by heating, incubate cells in PBS containing 300 mM NaCl at 65 °C for 6 h. Mix DNA solution with the same volume of phenol:chloroform:isoamyl solution and spin in a standard microcentrifuge (13,523 x g, room temperature) for 5 min. Transfer the upper aqueous phase into a fresh tube and repeat extraction. Add 1/10 volume of 3 M sodium acetate (pH 5.2) and 2.5 volumes of ethanol to the DNA solution and precipitate DNA at -20 °C for 30 min. Spin down the precipitate in a standard microcentrifuge (13,523 x g, 4 °C) for 15 min, carefully remove supernatant, and wash once with 70% ethanol. Air dry the pellet and dissolve in 50 μl TE buffer.
- To isolate mononucleosome-sized fragments, separate DNA on 2% agarose gels. Excise the ~150 bp band from the gel and purify DNA with a QIAquick Gel Extraction Kit according to manufacturer’s instruction. Elute in 30-50 μl elution buffer.
- Measure DNA concentration on a NanoDrop 2000 spectrophotometer and store at -20 °C. The yield of mononucleosomal DNA from a 10 cm-dish of NIH/3T3 cells is between 4-8 μg.
- Fix NIH/3T3 cells on a 10 cm-dish (80% confluency) by adding 270 μl of 37% formaldehyde solution to 10 ml culture medium (1% formaldehyde final) for 10 min at room temperature. Quench excessive formaldehyde by adding 1/20 volume of 2.5 M glycine solution (125 mM glycine final) for 5 min.
- Ligation of linkers to mononucleosomal DNA
- To generate blunted and phosphorylated DNA ends, set up the reaction as described below using the Quick Blunting Kit. Incubate at room temperature for 30 min followed by enzyme inactivation at 70 °C for 10 min.
10x blunting buffer 2.5 μl 1 mM dNTP 2.5 μl Enzyme mix 0.5 μl DNA 2.0 μg H2O to 25 μl - Dilute linkers S and L in TE buffer at a final concentration of 20 µM each (mass concentration of ~0.3 µg/µl). Anneal S and L by the following program on a PCR machine:
95 °C, 15 min
65 °C, 15 min (slope 0.01 °C/sec)
55 °C, 15 min (slope 0.01 °C/sec)
45 °C, 15 min (slope 0.01 °C/sec)
37 °C, 15 min (slope 0.01 °C/sec)
25 °C, 15 min (slope 0.01 °C/sec)
4 °C, 5 min - To ligate double-stranded linker L+S (from step B2) to mononucleosomal DNA (from step B1) set up the reaction as described below, and incubate the reaction at 16 °C overnight. Inactivate the enzyme at 65 °C for 20 min, purify the DNA with the QIAquick PCR Purification Kit and elute in 20 µl TE buffer.
10x ligation buffer 2 μl 100 mM ATP 1 μl T4 DNA ligase 1 μl DNA (from step B1) 1 μg Linker L+S (from step B2) 1 μg H2Oto 20 μl
- To generate blunted and phosphorylated DNA ends, set up the reaction as described below using the Quick Blunting Kit. Incubate at room temperature for 30 min followed by enzyme inactivation at 70 °C for 10 min.
- LM-PCR
- Label the PCR primer specific for the nucleosomal region of interest radioactively by setting up the reaction below and incubating for 1 h at 37 °C. After inactivating PNK at 65 °C for 20 min purify the primer with the QIAquick Nucleotide Removal Kit and elute in 20 µl TE buffer.
10x PNK buffer 2 μl PNK 3 μl [γ-32P]-ATP (3.3 µM) 9 μl Primer (20 μM) 1 μl H2O 8 μl - Set up LM-PCR reaction containing linker-ligated mononucleosomal DNA (from step B3), linker primer L and the 32P-labeled specific primer (from step C1) as follows:
GoTaq PCR Master Mix 7.5 μl DNA (from step B3) 2 μl Linker L (10 μM) 0.5 μl 32P-primer (from step C1) 2 μl DMSO 0.5 μl H2O 2.5 μl - Run the following ‘Touchdown’-PCR program:
Denaturation 94 °C 5 min
94 °C 30 sec, 65 °C 30 sec, 72 °C 30 sec; x 3 cycles
94 °C 30 sec, 64 °C 30 sec, 72 °C 30 sec; x 3 cycles
94 °C 30 sec, 63 °C 30 sec, 72 °C 30 sec; x 3 cycles
94 °C 30 sec, 62 °C 30 sec, 72 °C 30 sec; x 3 cycles
94 °C 30 sec, 61 °C 30 sec, 72 °C 30 sec; x 3 cycles
94 °C 30 sec, 60 °C 30 sec, 72 °C 30 sec; x 15 cycles
Complete extension 72 °C 10 min
- Label the PCR primer specific for the nucleosomal region of interest radioactively by setting up the reaction below and incubating for 1 h at 37 °C. After inactivating PNK at 65 °C for 20 min purify the primer with the QIAquick Nucleotide Removal Kit and elute in 20 µl TE buffer.
- Gel electrophoresis
- Prepare 6% denaturing urea-polyacrylamide gel (see Recipes).
- Assemble electrophoresis and pre-run the gel in 1x TBE for 30 min at 250 V. The pre-run allows equilibration of the gel/buffer system and heats up the gel to the operating temperature of 45-55 °C.
- After the pre-run, rinse gel pockets by flushing with fresh buffer using a syringe.
- Add an equal volume of formamide loading buffer to the LM-PCR reaction (from step C3) and incubate for 5 min at 95 °C, then quickly chill on ice. This step should be carried out during the pre-run of the gel so that the LM-PCR/loading buffer mix is on ice while rinsing the gel pockets. Thereby the time between pre-run and sample loading is minimized to avoid cooling down of the gel.
- Load 5 µl of the LM-PCR/loading buffer mix. Restart the electrophoresis and run for 1.5-2 h.
- Disassemble the gel set-up, put the gel on a Whatman paper and cover the opposite gel side with plastic wrap. It does not matter which side of the gel faces down. However, it is helpful to clip off one upper corner of the gel to locate its position after drying.
- Dry the gel at 80 °C for 1-2 h under vacuum.
- Expose gel to phosphor imaging plate overnight.
- Scan screen with phosphor imaging instrument and quantify bands representing different nucleosomal positions with appropriate software (e.g., AIDA Image Analyzer).
- Prepare 6% denaturing urea-polyacrylamide gel (see Recipes).
Data analysis
Detection of the radioactive signals from the LM-PCR products representing different nucleosome positions enables a relative, semi-quantitative analysis. Quantify the signal intensities with appropriate software and relate them within one sample/lane to each other. For instance, for the two nucleosome configurations at the rDNA promoter, NucU and NucD, we used the NucU/NucD ratio to compare changes between different physiological conditions (Zhao et al., 2016a and 2016b). Using the internal ratios of nucleosome positions for comparison rather than comparing the same positions between samples diminishes the effect of handling variations between samples and thus provides more robust results. Verify the reproducibility of the data by repeating the LM-PCR at least twice (technical replicates) and by performing the whole procedure at least three times with individually prepared nucleosomal DNA samples (biological replicates). Moreover, using a different gene-specific primer in the LM-PCR is also a good way to validate the results. If comparing internal ratios of nucleosome positions between two differently treated samples of cells, use a paired two-tailed Student’s t-test to calculate the statistical significance.
Recipes
- Permeabilization buffer
150 mM sucrose
80 mM KCl
35 mM HEPES (pH 7.4)
5 mM K2HPO4
5 mM MgCl2
0.5 mM CaCl2
0.05% L-α-lysophosphatidylcholine - MNase digestion buffer
150 mM sucrose
50 mM NaCl
50 mM Tris-HCl (pH 7.4)
2 mM CaCl2
50 U/ml MNase - TE buffer
20 mM Tris-HCl (pH 8.0)
2 mM EDTA - 10x TBE buffer (1 L)
108 g of Tris base
55 g of boric acid
40 ml of 0.5 M of EDTA (pH 8.0) - Formamide loading buffer
95% formamide
0.025% xylene cyanol
0.025% bromophenol blue
18 mM EDTA
0.025% SDS - Denaturing polyacrylamide gel (6%)

Acknowledgments
We thank Ingrid Grummt for comments on the manuscript. This work was funded by the Thuringian country program ProExzellenz (RegenerAging–FSU-I-03/14) of the Thuringian Ministry for Research (TMWWDG), Deutsche Forschungsgemeinschaft (SFB 1036), ‘CellNetworks’ (EcTop Survey 2014), and the Baden-Württemberg Stiftung.
The protocol was adapted from previous work (Li et al., 2006; Zhao et al., 2016a and 2016b).
References
- Bell O, Tiwari VK, Thoma NH, Schubeler D (2011). Determinants and dynamics of genome accessibility. Nat Rev Genet 12(8): 554-564
- Hughes, A. L. and Rando, O. J. (2014). Mechanisms underlying nucleosome positioning in vivo. Annu Rev Biophys 43: 41-63.
- Li, J., Langst, G. and Grummt, I. (2006). NoRC-dependent nucleosome positioning silences rRNA genes. EMBO J 25(24): 5735-5741.
- McPherson, C. E., Shim, E. Y., Friedman, D. S. and Zaret, K. S. (1993). An active tissue-specific enhancer and bound transcription factors existing in a precisely positioned nucleosomal array. Cell 75(2): 387-398.
- Soutoglou, E. and Talianidis, I. (2002). Coordination of PIC assembly and chromatin remodeling during differentiation-induced gene activation. Science 295(5561): 1901-1904.
- Tsompana, M. and Buck, M. J. (2014). Chromatin accessibility: a window into the genome. Epigenetics Chromatin 7(1): 33
- Xie, W., Ling, T., Zhou, Y., Feng, W., Zhu, Q., Stunnenberg, H. G., Grummt, I. and Tao, W. (2012). The chromatin remodeling complex NuRD establishes the poised state of rRNA genes characterized by bivalent histone modifications and altered nucleosome positions. Proc Natl Acad Sci U S A 109(21): 8161-8166.
- Zhao, Z., Dammert, M. A., Grummt, I. and Bierhoff, H. (2016a). lncRNA-induced nucleosome repositioning reinforces transcriptional repression of rRNA genes upon hypotonic stress. Cell Rep 14(8): 1876-1882.
- Zhao, Z., Dammert, M. A., Hoppe, S., Bierhoff, H. and Grummt, I. (2016b). Heat shock represses rRNA synthesis by inactivation of TIF-IA and lncRNA-dependent changes in nucleosome positioning. Nucleic Acids Res 44(17): 8144-8152.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Zhao, Z. and Bierhoff, H. (2017). Nucleosome Positioning Assay. Bio-protocol 7(10): e2285. DOI: 10.21769/BioProtoc.2285.
Category
Cancer Biology > General technique > Biochemical assays > DNA structure and alterations
Molecular Biology > DNA > DNA structure
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.