- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Ultradeep Pyrosequencing of Hepatitis C Virus to Define Evolutionary Phenotypes
Published: Vol 7, Iss 10, May 20, 2017 DOI: 10.21769/BioProtoc.2284 Views: 6854
Reviewed by: Yannick DebingRaju GhoshVamseedhar RayaproluAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






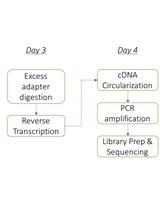

Abstract
Analysis of hypervariable regions (HVR) using pyrosequencing techniques is hampered by the ability of error correction algorithms to account for the heterogeneity of the variants present. Analysis of between-sample fluctuations to virome sub-populations, and detection of low frequency variants, are unreliable through the application of arbitrary frequency cut offs. Cumulatively this leads to an underestimation of genetic diversity. In the following technique we describe the analysis of Hepatitis C virus (HCV) HVR1 which includes the E1/E2 glycoprotein gene junction. This procedure describes the evolution of HCV in a treatment naïve environment, from 10 samples collected over 10 years, using ultradeep pyrosequencing (UDPS) performed on the Roche GS FLX titanium platform (Palmer et al., 2014). Initial clonal analysis of serum samples was used to inform downstream error correction algorithms that allowed for a greater sequence depth to be reached. PCR amplification of this region has been tested for HCV genotypes 1, 2, 3 and 4.
Keywords: Ultradeep pyrosequencingBackground
Analysis of UDPS datasets derived from virus amplicons frequently relies on software tools that are not optimized for amplicon analysis, assume random incorporation of sequencing mutations and are focused on finding true sequences rather than false variants. These difficulties are further complicated by the presence of hypervariable regions present in RNA virus genomes. Many studies utilizing UDPS look to overcome these issues by applying arbitrary frequency cut offs to the data, resulting in the loss of minor variants. Here, a temporally matched clonal dataset, together with an error correction methodology designed to overcome the problems outlined, facilitated the retention of valuable sequence information.
Materials and Reagents
- 1.5 ml tube (SARSTEDT, catalog number: 72.690.001 )
- 200 µl MicroAmp® PCR tube (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: N8010840 )
- Clean stainless steel blade
- One Shot® TOP10 Competent Cells (Thermo Fisher Scientific, InvitrogenTM, catalog number: C404003 )
- QIAamp® Viral RNA mini kit (QIAGEN, catalog number: 52904 )
- Random primer (Promega, catalog number: C1181 )
- Deoxynucleoside triphosphate (dNTP’s, 100 mM) set, PCR grade (Roche Molecular Systems, catalog number: 11969064001 )
- AMV reverse transcriptase (Promega, catalog number: M5101 )
- RNasin® Ribonuclease inhibitor (Promega, catalog number: N2511 )
- Outer-forward primer: 5’- ATGGCATGGGATATGAT -3’ (10 pmol/µl, Eurofins)
- Outer-reverse primer: 5’- AAGGCCGTCCTGTTGA -3’ (10 pmol/µl, Eurofins)
- Inner-forward primer: 5’- GCATGGGATATGATGATGAA -3’ (10 pmol/µl, Eurofins)
- Inner-reverse primer: 5’- GTCCTGTTGATGTGCCA -3’ (10 pmol/µl, Eurofins)
- Pwo DNA polymerase (5 U/µl,) including 10x reaction buffer (- MgSO4) and MgSO4 stock solution (25 mM) (Roche Molecular Systems, catalog number: 11644955001 )
- dH2O (Sigma-Aldrich, catalog number: W4502 )
- Sybr safe DNA gel stain (Thermo Fisher Scientific, InvitrogenTM, catalog number: S33102 )
- Agarose (Sigma-Aldrich, catalog number: A9539 )
- GeneRuler 100 bp Plus DNA ladder (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: SM0323 )
- Gel extraction kit (QIAGEN, catalog number: 28704 )
- CloneJet PCR Cloning Kit (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: K1231 )
- GeneJet Plasmid Miniprep Kit (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: K0503 )
- Trizma® base (Sigma-Aldrich, catalog number: T1503 )
- Acetic acid glacial (BDH Laboratory Supplies, catalog number: 10001CU )
- Ethylenediaminetetraacetic acid solution 0.5 M (EDTA) (Sigma-Aldrich, catalog number: 03690 )
- 1x TAE (see Recipes)
Equipment
- PCR thermal cycler (Thermo Fisher Scientific, Applied BiosystemsTM, model: Applied Biosystems® 2720 )
- BioPhotometer (Eppendorf, http://arboretum.harvard.edu/wp-content/uploads/Biophotometer-manual.pdf)
- Water bath (JULABO, model: SW22 )
- Orbital shaker incubator (Grant, model: ES-80 )
- Ultraviolet transilluminator (UVP, model: TMW-20 )
Software
- SFFFile tools (Roche Molecular Systems)
- k-mer error correction (KEC) and empirical threshold (ET) (Skums et al., 2012)
- MEGA 6.0 (Tamura et al., 2013)
Procedure
- RNA extraction and cDNA generation
- Whole patient serum, surplus to diagnostic testing requirements and with a mean viral titer of 6 HCV RNA log10 IU/ml, was used as the starting material.
- RNA was extracted from 140 µl of serum using QIAamp® Viral RNA mini kit according to the manufacturer’s instructions into 1.5 ml RNase free tubes and a final volume of 50 µl.
- 11 µl of extracted viral RNA was mixed with 1 µl (0.5 µg) random primer.
- Samples were incubated at 75 °C for 10 min.
- To this was added a master mix which contained 2 µl (80 mM) dNTP mix, 1 µl (10 U) AMV reverse transcriptase, 1 µl (40 U) RNasin, 4 µl AMV reaction buffer.
- cDNA generation took place at 42 °C for 60 min, followed by 94 °C for 3 min.
- Samples were kept at 4 °C until required.
- Whole patient serum, surplus to diagnostic testing requirements and with a mean viral titer of 6 HCV RNA log10 IU/ml, was used as the starting material.
- Nested PCR to amplify the HCV E1/E2 gene junction
- Prepare the primary PCR master mix to a final volume of 45 µl:
Outer-forward primer: 1.5 µl Outer-reverse primer: 1.5 µl 10x reaction buffer (- MgSO4): 5 µl dNTP mix: 1 µl MgSO4 stock solution: 3 µl Pwo: 0.5 µl PCR grade water: 32.5 µl - 5 µl of cDNA is then added to the master mix.
- 1° PCR cycle parameters:
- Initial denaturation: 3 min at 94 °C
- Cycle conditions (repeat for 35 cycles):
Denaturation: 15 sec at 94 °C
Annealing: 30 sec at 51 °C
Extension: 30 sec at 72 °C - Final extension: 7 min at 72 °C
- Initial denaturation: 3 min at 94 °C
- Keep sample at 4 °C until required.
- Prepare master mix for secondary PCR to a final volume of 46 µl:
Inner-forward primer: 1.5 µl Inner-reverse primer: 1.5 µl 10x reaction buffer (- MgSO4): 5 µl dNTP mix: 1 µl MgSO4 stock solution: 2 µl Pwo: 0.5 µl PCR grade water: 34.5 µl - 4 µl of primary PCR sample is then added to the master mix.
- 2° PCR cycle parameters:
- Initial denaturation: 3 min at 94 °C
- Cycle conditions (repeat for 35 cycles):
Denaturation: 15 sec at 94 °C
Annealing: 30 sec at 53 °C
Extension: 30 sec at 72 °C - Final extension: 7 min at 72 °C
- Initial denaturation: 3 min at 94 °C
- Samples were kept at 4 °C until required.
- To ensure that the initial amount of the template was not limiting, 1:100 dilution of the viral RNA was prepared which, when used as the starting template for nested PCR as described, should yield an amplicon visualized by gel electrophoresis for each sample.
- Prepare the primary PCR master mix to a final volume of 45 µl:
- Preparation of samples for pyrosequencing
- Two 2% TAE agarose gels were poured, one containing Sybr safe DNA gel stain and one without.
- Once set, the gels were split in two, with one half of the gel containing the gel stain joined with the second gel without gel stain.
- The 50 µl amplicon sample was split in two (10 µl and 40 µl) and resolved on the above gel. The 10 µl sample was stained using Sybr safe, while the 40 µl sample was not stained and went forward for downstream procedures. The resultant amplicon in this instance was 320 bp (Figure 1).
- The region of the gel containing the unstained band (40 µl sample) was cut out using a clean stainless steel blade using the stained 10 µl sample as a positioning guide and transferred to a clean 1.5 ml tube.
- The amplicon was gel extracted using a gel extraction kit according to the manufacturer’s instructions.
- Extracted amplicons were quantified using a BioPhotometer.
- Samples were prepared in equimolar concentrations and diluted to a final concentration of 1 x 107 molecules/ml.
- Pyrosequencing was outsourced to Roche 454 Life Sciences (Brandford, CT, USA).
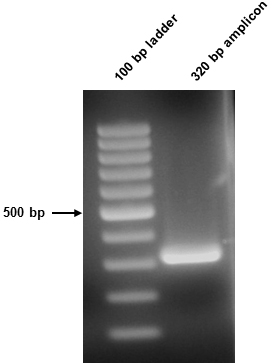
Figure 1. Amplicon visualization. Successful amplification of the 320 bp amplicon was confirmed following agarose gel electrophoresis. 10 µl of the 2° PCR sample was loaded.
- Two 2% TAE agarose gels were poured, one containing Sybr safe DNA gel stain and one without.
- Clonal analysis
- Purified amplicons were cloned using CloneJet PCR Cloning Kit and transformed into One Shot® TOP10 Competent Cells using the manufacturer’s instructions using a molar ratio of 3:1 insert to vector.
- 20 clones per sample were generated.
- Plasmids were purified using GeneJet Plasmid Miniprep Kit as per manufacturer’s instructions.
- Sequencing of E1/E2 inserts was performed by Eurofins.
- All trace files were inspected to exclude sequences where double peaks or regions of ambiguous sequence were present.
- Purified amplicons were cloned using CloneJet PCR Cloning Kit and transformed into One Shot® TOP10 Competent Cells using the manufacturer’s instructions using a molar ratio of 3:1 insert to vector.
- Data handling and error correction
- The raw sff data files were managed using SFFFile tools.
- Low-quality reads and reads shorter than 90% of the expected amplicon lengths were removed.
- Phylogenetic separation of the clonal data using a general time-reversible model with gamma-distributed and invariant sites (GTR+G+I) using MEGA 6.0 (Tamura et al., 2013).
- Main branches with bootstrap values (of 1,000 resamplings) > 85 were categorised as (sub-)lineages (Palmer et al., 2014).
- Two 24-bp motifs, that defined the HVR1 amino acid profile of each (sub-)lineage, were subsequently applied to the sequence analysis pipeline. The first 15-bp of the motif span the conserved 3’-end of E1. The remaining 9-bp include the first three amino acids of the HVR1 at the 5’-end of E2.
- The overall number of motifs used reflected the observed changes in the dominant HVR1 over time. For each (sub-)lineage, two motif reference sequences were deemed sufficient.
- To increase the sensitivity of the sequencing error correction algorithms (KEC-ET), the UDPS data was partitioned according to the presence of corresponding motifs.
- In order to ensure the quality of the analyzed data and the absence of PCR and sequencing chimeras, reads that had more than a 3 bp difference from the best-matching sequence from this motif set were removed.
- KEC consists of the three stages
- In stage 1, the set of k-mers (substring of fixed length k) of reads from the processed data set is calculated and the distribution of frequencies of k-mers is analyzed. The error threshold is calculated as the minimal frequency of k-mers separating two different distributions.
- In stage 2, k-mers with frequencies lower than the error threshold are considered erroneous and are used to identify and correct the errors. The corrections are based on an analysis of different factors, including the length of a segment of consecutive erroneous k-mers, the sequences of nucleotides at the end of that segment, and the frequencies of the similar correct k-mers. The procedure of error correction is repeated iteratively i times.
- In stage 3, the reads containing k-mers that were not corrected in stage 2 are discarded.
- The following parameters of KEC were used: k = 25 and i = 3.
- The raw sff data files were managed using SFFFile tools.
Data analysis
A more complete description of the data handling and error correction procedure can be found in the original article, http://jvi.asm.org/content/88/23/13709.short (Palmer et al., 2014).
Notes
All serum samples were genotyped and quantified by the Molecular Virology Diagnostic & Research Laboratory at Cork University Hospital, Cork, Ireland. https://www.ucc.ie/en/meddept/people/liam-fanning/mvdrl/
Recipes
- 1x TAE
4.84 g Tris base
1.15 ml acetic acid glacial
2 ml 0.5 M EDTA
Add dH2O to 1 L
References
- Palmer, B. A., Dimitrova, Z., Skums, P., Crosbie, O., Kenny-Walsh, E. and Fanning, L. J. (2014). Analysis of the evolution and structure of a complex intrahost viral population in chronic hepatitis C virus mapped by ultradeep pyrosequencing. J Virol 88(23): 13709-13721.
- Skums, P., Dimitrova, Z., Campo, D. S., Vaughan, G., Rossi, L., Forbi, J. C., Yokosawa, J., Zelikovsky, A. and Khudyakov, Y. (2012). Efficient error correction for next-generation sequencing of viral amplicons. BMC Bioinformatics 13 Suppl 10: S6.
- Tamura, K., Stecher, G., Peterson, D., Filipski, A. and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12): 2725-2729.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Palmer, B. A., Dimitrova, Z., Skums, P., Crosbie, O., Kenny-Walsh, E. and Fanning, L. J. (2017). Ultradeep Pyrosequencing of Hepatitis C Virus to Define Evolutionary Phenotypes. Bio-protocol 7(10): e2284. DOI: 10.21769/BioProtoc.2284.
Category
Molecular Biology > RNA > RNA sequencing
Microbiology > Microbial genetics > RNA > Sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.