- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Virtual Screening of Transmembrane Serine Protease Inhibitors
(*contributed equally to this work) Published: Vol 7, Iss 8, Apr 20, 2017 DOI: 10.21769/BioProtoc.2246 Views: 9994
Reviewed by: HongLok LungShyam SrivatsTim Andrew Davies SmithVanesa Olivares-Illana
Original research article
The authors used this protocol in:

Apr 2016

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The human family of type II transmembrane serine proteases includes 17 members. The defining features of these proteases are an N-terminal transmembrane domain and a C-terminal serine protease of the chymotrypsin (S1) fold, separated from each other by a variable stem region. Recently accumulated evidence suggests a critical role for these proteases in development of cancer and metastatic capacity. Both the cancer relevance and the accessibility of the extracellularly oriented catalytic domain for therapeutic and imaging agents have fueled drug discovery interest in the type II class of transmembrane serine proteases. Typically, the initial hit discovery processes aim to identify molecules with verifiable activity at the drug target and with sufficient drug-like characters. We present here protocols for structure-based virtual screening of candidate ligands for transmembrane serine protease hepsin. The methods describe use of the 3D structure of the catalytic site of hepsin for molecular docking with ZINC, which is a molecular database of > 30 million purchasable compounds. Small candidate subsets were experimentally tested with demonstrable hits, which provided meaningful cues of the ligand structures for further lead development.
Keywords: Molecular modelingBackground
Controlled proteolytic activity plays a fundamental role in cellular processes and signaling, as evidenced by the presence of proteases in all organisms, including viruses, prokaryotes and eukaryotes. Not surprisingly, aberrantly regulated protease activity is causal to wide variety of human pathologies such as cardiovascular and inflammatory diseases, osteoporosis, neurological disorders and cancer (Turk, 2006; Bachovchin and Cravatt, 2012). In particular, development of primary cancer and metastatic capacity has been linked to several different classes of proteases including matrix metalloproteinases (MMPs), cysteine proteases (cathepsins) and membrane-associated serine proteases (Lopez-Otin and Matrisian, 2007). Recent clinical, genetic and functional data, suggesting a critical role for membrane-associated serine proteases in solid cancers, including cancer of prostate, ovarian and breast, have prompted new interest in the development of small molecule serine protease inhibitors for the treatment of cancer. Hepsin is a type II transmembrane serine protease and an attractive target for serine protease drug development due to frequent hepsin overexpression in common solid cancers, such as prostate and breast cancer, confinement of its overexpression on the membranes of cancer cells and due to positioning of the catalytic domain to the extracellular, i.e., more reachable, side of the cells (Antalis et al., 2010).
We describe here protocols for structure-based virtual screening of serine protease inhibitors using the catalytic site of hepsin for docking with drug-like subset of ZINC database. On the basis of virtual screening results, altogether 24 candidate compounds were purchased for further biochemical validation. Cell-free ELISA-based fluorogenic enzymatic assay using recombinant hepsin and fluorogenic peptide substrate Boc-Gln-Arg-Arg-AMC (BACHEM) was used for experimental validation and 30% inhibition of peptidolytic activity was set as threshold. Cut-off value was based on a notion that with the initial high micromolar concentration of compounds more than 30% inhibition would be required to determine reasonable IC50 value (Goswami et al., 2015; Tervonen et al., 2016) (Figures 1A and 1B). With these criteria, 3 out of 24 tested compounds showed inhibition potency (Tervonen et al., 2016). In conclusion, even though structure-based virtual screening is often considered as a complementary drug screening approach, the protocols reported here allowed us to demonstrate the feasibility and provided meaningful structural scaffolds for further development of specific serine protease inhibitors. However, it is important to stress that virtual screening hits typically do not demonstrate high potency, which is also true to the present screen. High-affinity ligands typically become available only after skillful medicinal chemistry optimization of selected hit structures.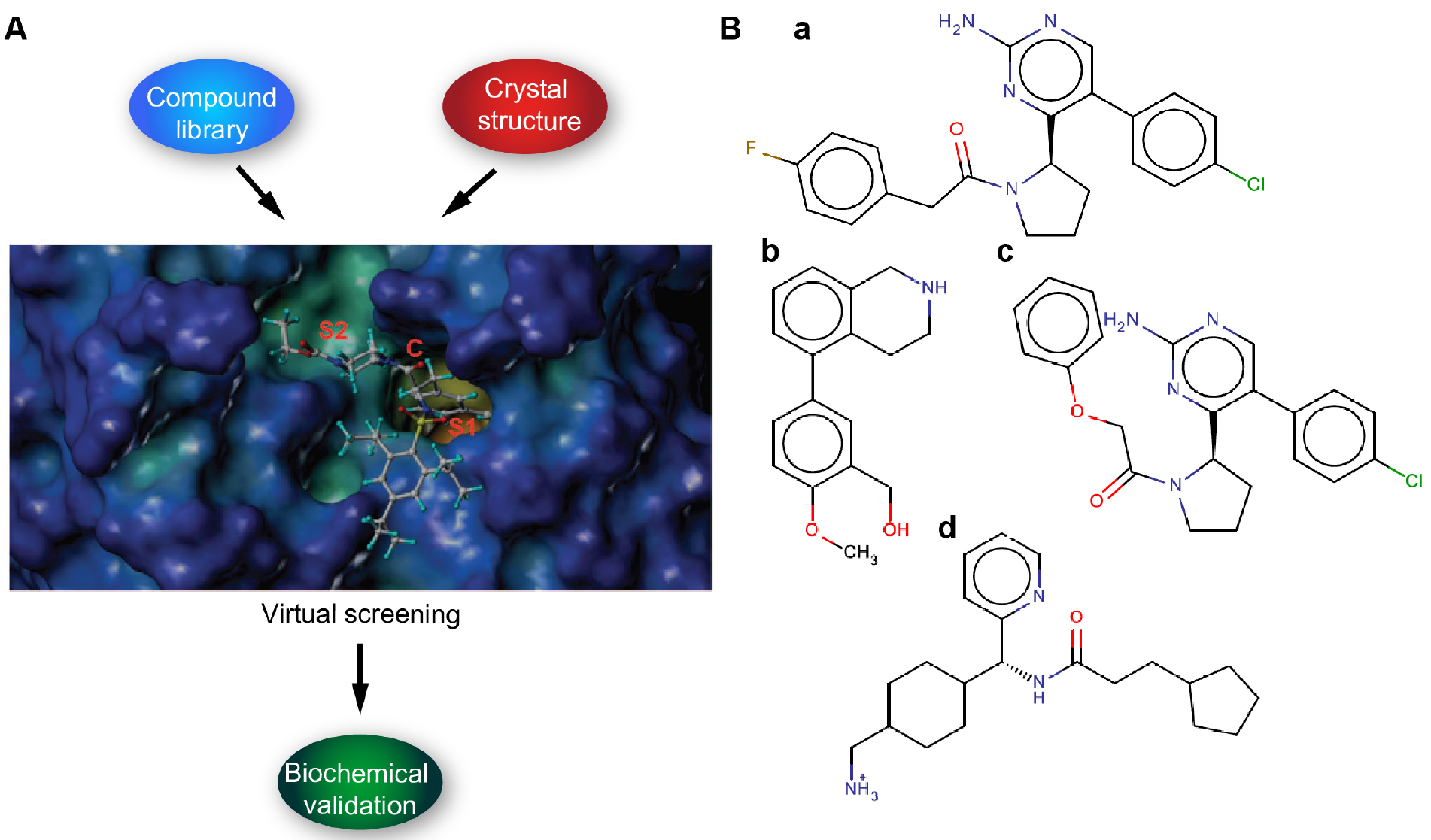
Figure 1. Schematic figure illustrating the workflow. A. Virtual screening (compound) library is prepared and the target protein structure analyzed and prepared. The virtual screening is carried out by docking (Glide SP/XP) and the putative binders are validated by in vitro biochemical assays. B. Examples of candidate ligands (a-d).
The virtual screening protocol presented here is a modified version from the original protocols described in our recent research paper (Tervonen et al., 2016). The modifications make the protocol fully compatible with the most updated versions of ZINC database and Schrödinger software. While the current virtual screening protocol is tailored for users of Schrödinger software and ZINC database, the protocol is versatile and also compatible with other molecular modeling packages. However, implementation of the present virtual screening protocol with other modeling packages would require a careful platform-specific validation of the docking settings. As a guidance, we provide a general overview of the validation procedures used in the present screen.
Materials and Reagents
- ViewPlate-96 black assay plates (PerkinElmer, catalog number: 6005225 )
- Recombinant human hepsin protein (R&D Systems, catalog number: 4776-SE-010 )
- DMSO (Sigma-Aldrich, catalog number: D8418 )
- Fluorogenic peptide substrate Boc-Gln-Arg-Arg-AMC (Bachem, catalog number: I-1655.0025 )
- Small-molecule compounds were purchased either from Asinex Corporation (Winston-Salem, NC, http://www.asinex.com) or MolPort (Riga, Latvia, https://www.molport.com/shop/index)
- WX-UK1 was a kind gift from Dr. Ramachandra (Aurigene Discovery Technologies Limited, Bangalore, India), however, it is also commercially available (for example, AURUM Pharmatech, catalog number: Z-3200 )
- Trizma base (Tris) (Sigma-Aldrich catalog number: T1503 )
- Calcium chloride (CaCl2) (Sigma-Aldrich, catalog number: 499609 )
- Brij-35 (Brij L23 30% solution in H2O) (Sigma-Aldrich, catalog number: B4184 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: 31434-M )
- Activation buffer for recombinant human hepsin protein (see Recipes)
- Assay buffer for recombinant human hepsin protein (see Recipes)
Equipment
- ELISA plate reader FLUOstar Omega (BMG LABTECH, model: FLUOstar Omega )
- Computer hardware provided by CSC - IT Center for Science Ltd (Espoo, Finland)
Software
- Modeling software
Schrödinger Suite (Schrödinger, LLC, New York, NY) with Protein Preparation Wizard (Sastry et al., 2013), LigPrep, MacroModel and Glide (Friesner et al., 2004 and 2006; Halgren et al., 2004) modules, and a protocol version 2016-2 (https://www.schrodinger.com/suites/small-molecule-drug-discovery-suite) - Databases
- Screening and validation: ZINC database (downloadable via http://zinc15.docking.org/). ZINC database includes over 100 million purchasable compounds in 3D formats (Sterling and Irwin, 2015)
- Protein structure database (http://www.rcsb.org/pdb/home/home.do)
Procedure
- Preparation of the target protein structure for virtual screening
The hepsin protein structure (PDB id 1O5E, from protein structure database, www.rcsb.org, see ‘Online tools’) can be automatically downloaded with Protein Preparation Wizard of Schrödinger suite using ‘biological unit’-option. With other types of softwares one should use the manual downloading and read in the structure by software preferred method. The pre-processing is carried out with default methods except keeping all the water molecules. H-bond refinement should be carried out with default pH value 7. All water molecules with at least 3 H-bonds with non-water atoms are saved and the whole structure is minimized with default settings with heavy atoms converged at RMSD 0.3Å. The Glide Grid file is created using the above mentioned structure and the center of the Grid was at Tyr94 (Figure 2). The center of the GRID is referring to the center of molecular field calculation during the GRID creation. Hydroxyl groups of Tyr94, Tyr146, Ser195 and Tyr228 are allowed to rotate for Grid generation. Addition of free rotatable bonds doubles the Glide calculation time and should be limited to keep the computational time at reasonable level. Also, it is strongly recommended that the Glide Grid file will be constructed so that OPLS3 force field is used, if possible. If another software is used, one should use the corresponding protein preparation method. The critical point is to make sure that the structural quality of the protein is checked, proper ionization status are fixed for polar sidechains and right rotamers for (pseudo)symetric residues are used.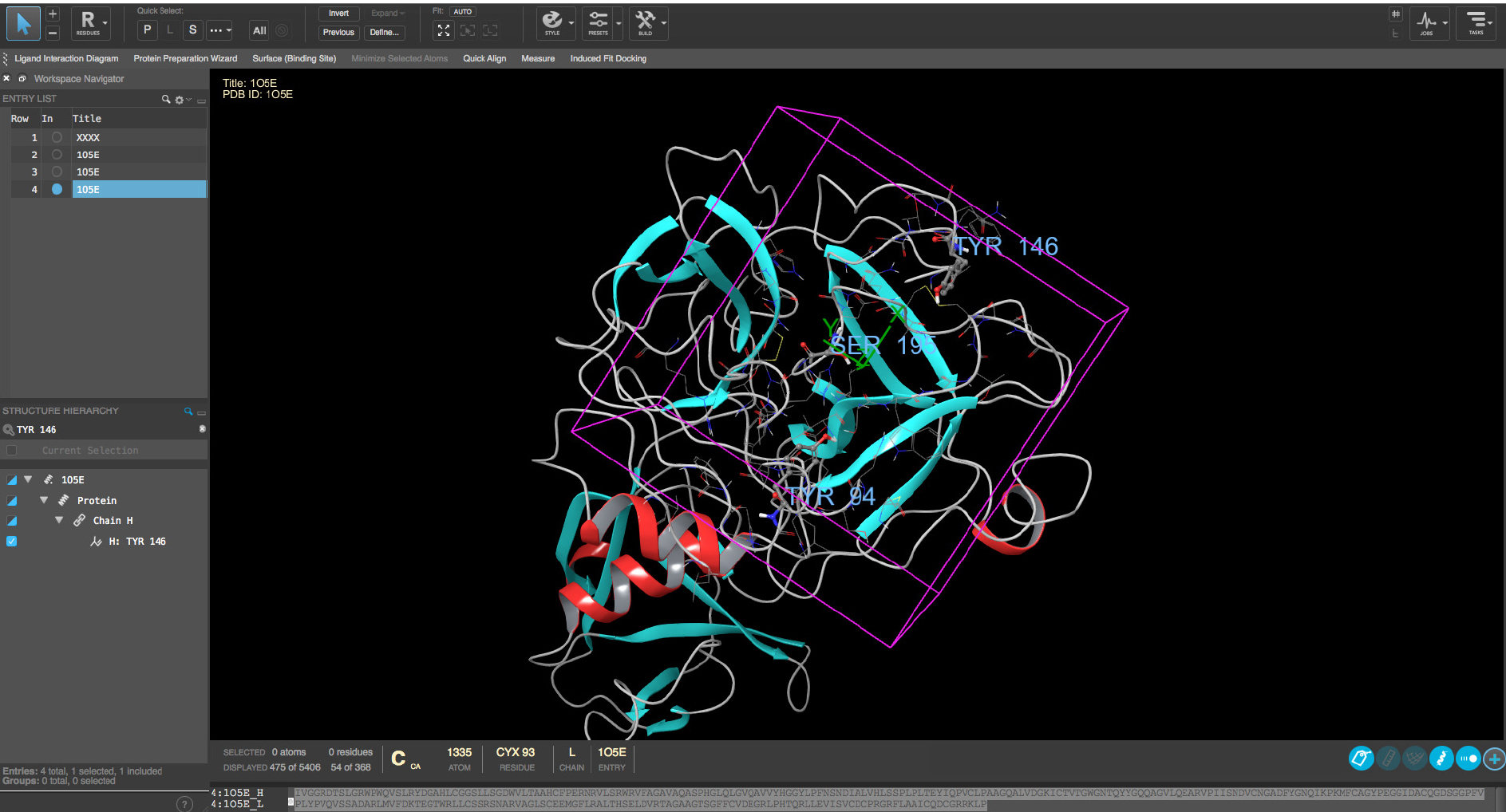
Figure 2. Definition of GRID region for docking. The center of the GRID is based on the location of residues TYR94, TYR146 and SER195 (residues marked with labels) and the GRIF region is shown with magenta box. - Preparation of the virtual screening compound library
The virtual screening docking library must be prepared using the methods and settings recommended by the authors of the virtual screening software. When choosing the virtual compound library, one should take into account the number of compounds intended for screening, possibilities for own synthesis and if the resulting hits are intended to be used as such or further modified in subsequent medicinal chemistry programs. We use the drug-like subset of ZINC library, downloaded as a SDF-file from the ZINC web page. SDF-file format is supported by all the major software brands. The SDF-file is prepared by Schrödinger LigPrep module with default settings (Figure 3). In short the preparation includes checking of 2D structure quality, fast 2D-3D transformation, analysis and modification of protonation state, modification of all reachable tautomers and further minimization of resulting molecules. In a typical case each molecule will be represented with 2-4 different ionization/protomer/tautomer states. For other types of software other than the Schrödinger software, we recommend ligand setup via LigPrep. In our own experience, the benefit of this method is that it yields high quality predictions for both ionization status (i.e., pKa) and also for tautomers. The prediction of tautomers option is not included in most of the other software packages.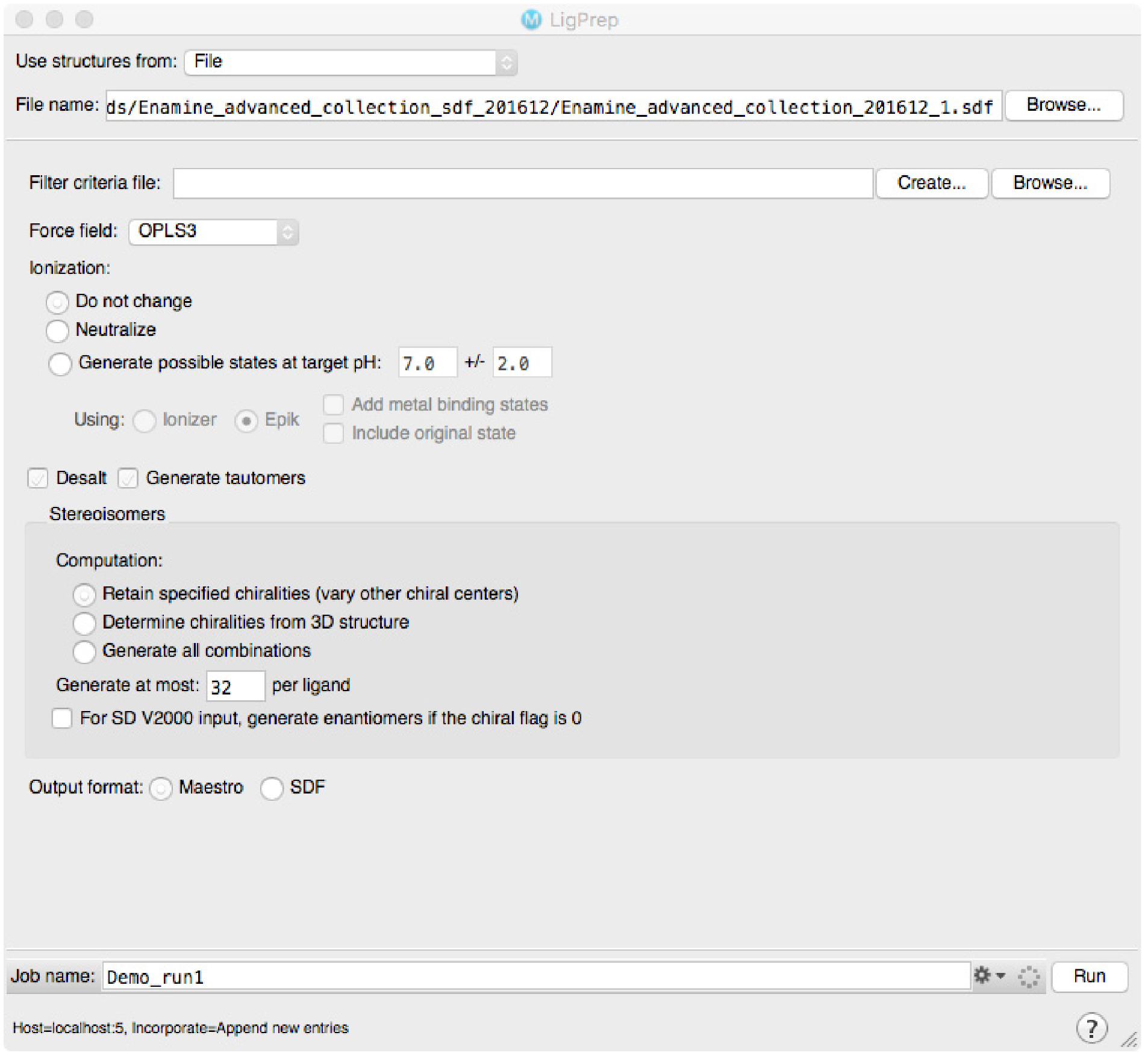
Figure 3. LigPrep window under Schrödinger Suite. The library for virtual screening is prepared according to the settings selected; the shown setting are the default ones in our protocol. - Virtual screening and analysis of the results
In our study (Tervonen et al., 2016) virtual screening was carried out as follows:- SP-docking is performed using the above-defined Grid file and the best 10% of compounds are further redocked with XP settings.
- In the first stage VHTS-settings were used and then SP settings were utilized only for the best 50,000-100,000 compounds (ranked according to the Glide-scoring function) and further, for the best 10%, XP-settings.
- In the original study OPLS2005 force field was used. However, we now strongly recommend OPLS3 force field, since it gives much better results compared to the original method.
- Clustering compounds was initially performed by visually studying the resulting docking poses.
- The aim of the visual inspection was to make sure that only reasonable docking poses are accepted. Typically some 10-20% of docking poses are unrealistic due to missing H-bonding contacts or unrealistic ligand conformations and these can be effectively screened by visual inspection.
- Compounds were then clustered by using the Interaction Fingerprint script available within Schrödinger.
- This script analyzed each of the docking pose based on the interaction profile (interaction fingerprints) and the poses are clustered by using those profiles. In our study we used the optimal number of clusters as evaluated by the script with default linkage method. For a detailed description of the method and its uses, see Deng et al. (2004) and Singh et al. (2006).
- As a result 24 clusters were created.
- In each cluster the most representative compounds are located in the center of the cluster. Those compounds are selected for in vitro assays.
- Validation of screen results by in vitro cell-free peptide cleavage assay for hepsin enzymatic activity protocols
- Recombinant human hepsin (rhHepsin) is diluted to 100 µg/ml in activation buffer.
- rhHepsin is activated by incubating the solution at 37 °C for 24 h.
- rhHepsin is diluted into 0.1 nM solution with assay buffer just before use.
- Small-molecule compounds are dissolved in DMSO to give 10 mM stock solution.
- The assay is performed in assay buffer in a 96-well plate.
- 0.1 nM final concentration of rhHepsin and 10 µM final concentration of small-molecule compound (and equal volume of DMSO control) in 100 µl reaction volume are incubated for 30 min at room temperature. Assay buffer is used as blank control.
- The reaction is started by adding a final concentration of 30 µM peptide substrate BOC-Gln-Arg-Arg-AMC.
- The plate is analyzed with ELISA plate reader by 350 nm excitation and 450 nm emission capture at room temperature.
- The inhibition % is determined by using the following formula:
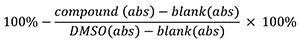
- Recombinant human hepsin (rhHepsin) is diluted to 100 µg/ml in activation buffer.
Data analysis
The described virtual screening protocol resulted in hundreds of potential hits. Using the protocols described above and in Tervonen et al. (2016), we performed an initial in vitro cell-free validation. More than 30% reduction in peptide cleavage relative to DMSO control with three technical replicates and two repeats was considered as a cut-off for hits. We ordered and tested 24 compounds identified by virtual screening as potential hits (Figure 1B) and few compounds made it close to the cut-off value (see Tervonen et al. [2016]). These compounds were used as skeletons for the next round of virtual screening, which is a reiterative process. In the study Tervonen et al. (2016) we chose to use WX-UK1, which was identified in parallel screen, because its binding pose against hepsin and superb inhibition efficiency in comparison to other tested molecules.
Notes
- Computer hardware
In addition to listed hardware above, we note that any high-end PC computer with any of the major operating system (Windows, Mac or Linux) and with either Intel or AMD processors is suitable for the virtual screening job but the process will then require more time. - Small-molecule compounds
We note that while the virtual screening stage of the project was low-cost and non-time consuming, one bottleneck in the screening was the price of the compounds. The typical price for commercially available compounds ranges from 50 € to 200 € per mg. Thus, screening of hundreds of compounds in cell-free assays would be the most expensive stage of the project. Also, the fact that most of the compounds are sold with minimum 1 mg pack size (synthesis limit) and that the initial cell-free screening only requires fraction of that amount makes purchasing of vast amount of compounds unreasonable. - Feasibility of virtual screening In our experience
Virtual screening as a method for identification of molecular tool compounds performed well in finding a small set of skeleton compounds even though only relatively small number of compounds identified as hits by molecular modeling were eventually tested in vitro (cell free assays with recombinant hepsin). These results indicate that virtual screening is in principle a feasible method for identification of transmembrane serine protease inhibitors but it would be advisable to use virtual screening in combination with other approaches. For example, starting with skeleton compounds with previously established inhibitory action against the transmembrane serine protease of interest or against closely related serine protease. The virtual screening strategy described here would be greatly facilitated by a possibility to purchase just a small microgram quantity of the candidate molecule instead of a milligram. There would be a significant market for any contract research laboratory able to miniaturize compound synthesis for purpose of testing significant number of virtual screening hits with low-cost.
Recipes
- Activation buffer for recombinant human hepsin (pH 8.0)
0.1 M Tris
10 mM CaCl2
0.15 M NaCl
0.05% Brij-35 - Assay buffer for recombinant human hepsin (pH 9.0)
50 mM Tris
Acknowledgments
Protocol is adapted from Tervonen et al. (2016). This study was funded by the Academy of Finland, Sigrid Jusélius Foundation, the Finnish Cancer Society, the Research Funds of the Helsinki University Central Hospital, Jane and Aatos Erkko Foundation, and Helsinki Graduate Program in Biotechnology and Molecular Biology and Innovative Medicines Initiative Joint Undertaking under grant agreement No.115188.
References
- Antalis, T. M., Buzza, M. S., Hodge, K. M., Hooper, J. D. and Netzel-Arnett, S. (2010). The cutting edge: membrane-anchored serine protease activities in the pericellular microenvironment. Biochem J 428(3): 325-346.
- Bachovchin, D. A. and Cravatt, B. F. (2012). The pharmacological landscape and therapeutic potential of serine hydrolases. Nat Rev Drug Discov 11(1): 52-68.
- Deng, Z., Chuaqui, C. and Singh, J. (2004). Structural interaction fingerprint (SIFt): A novel method for analyzing three-dimensionla protein-ligand binding interactions. J Med Chem 47(2): 337-344.
- Friesner, R. A., Banks, J. L., Murphy, R. B., Halgren, T. A., Klicic, J. J., Mainz, D. T., Repasky, M. P., Knoll, E. H., Shelley, M., Perry, J. K., Shaw, D. E., Francis, P. and Shenkin, P. S. (2004). Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47(7): 1739-1749.
- Friesner, R. A., Murphy, R. B., Repasky, M. P., Frye, L. L., Greenwood, J. R., Halgren, T. A., Sanschagrin, P. C. and Mainz, D. T. (2006). Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49(21): 6177-6196.
- Goswami, R., Wohlfahrt, G., Törmäkangas, O., Moilanen, A., Lakshminarasimhan, A., Nagaraj, J., Arumugam, K. N., Mukherjee, S., Chacko, A. R., Krishnamurthy, N. R., Jaleel, M., Palakurthy, R. K., Samiulla, D. S. and Ramachandra, M. (2015). Structure-guided discovery of 2-aryl/pyridin-2-yl-1H-indole derivatives as potent and selective hepsin inhibitors. Bioorg Med Chem Lett 25(22): 5309-5314.
- Halgren, T. A., Murphy, R. B., Friesner, R. A., Beard, H. S., Frye, L. L., Pollard, W. T. and Banks, J. L. (2004). Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 47(7): 1750-1759.
- Lopez-Otin, C. and Matrisian, L. M. (2007). Emerging roles of proteases in tumour suppression. Nat Rev Cancer 7(10): 800-808.
- Sastry, G. M., Adzhigirey, M., Day, T., Annabhimoju, R. and Sherman, W. (2013). Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27(3): 221-234.
- Singh, J.,Deng, Z., Narale, G. and Chuaqui, C. (2006). Structural interaction fingerprints: a new approach to organizing, mining, analyzing, and designing protein-small molecule complexes. Chem Biol Drug Des 67(1): 5-12.
- Sterling, T. and Irwin, J.J. (2015). ZINC 15--Ligand discovery for everyone. J Chem Inf Model 55(11): 2324-2337.
- Tervonen, T. A., Belitskin, D., Pant, S. M., Englund, J. I., Marques, E., Ala-Hongisto, H., Nevalaita, L., Sihto, H., Heikkila, P., Leidenius, M., Hewitson, K., Ramachandra, M., Moilanen, A., Joensuu, H., Kovanen, P. E., Poso, A. and Klefstrom, J. (2016). Deregulated hepsin protease activity confers oncogenicity by concomitantly augmenting HGF/MET signalling and disrupting epithelial cohesion. Oncogene 35(14): 1832-1846.
- Turk, B. (2006). Targeting proteases: successes, failures and future prospects. Nat Rev Drug Discov 5(9): 785-799.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Poso, A., Tervonen, T. and Klefström, J. (2017). Virtual Screening of Transmembrane Serine Protease Inhibitors. Bio-protocol 7(8): e2246. DOI: 10.21769/BioProtoc.2246.
Category
Cancer Biology > Cancer biochemistry > Protein
Biochemistry > Protein > Activity
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.