- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Efficient Generation of Multi-gene Knockout Cell Lines and Patient-derived Xenografts Using Multi-colored Lenti-CRISPR-Cas9


(*contributed equally to this work) Published: Vol 7, Iss 7, Apr 5, 2017 DOI: 10.21769/BioProtoc.2222 Views: 21446
Reviewed by: Gal HaimovichAnnis Elizabeth RichardsonAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
CRISPR-Cas9 based knockout strategies are increasingly used to analyze gene function. However, redundancies and overlapping functions in biological signaling pathways can call for generating multi-gene knockout cells, which remains a relatively laborious process. Here we detail the application of multi-color LentiCRISPR vectors to simultaneously generate single and multiple knockouts in human cells. We provide a complete protocol, including guide RNA design, LentiCRISPR cloning, viral production and transduction, as well as strategies for sorting and screening knockout cells. The validity of the process is demonstrated by the simultaneous deletion of up to four programmed cell death mediators in leukemic cell lines and patient-derived acute lymphoblastic leukemia xenografts, in which single cell cloning is not feasible. This protocol enables any lab with access to basic cellular biology equipment, a biosafety level 2 facility and fluorescence-activated cell sorting capabilities to generate single and multi-gene knockout cell lines or primary cells efficiently within one month.
Keywords: CRISPRBackground
Starting with curious initial observations of genetic elements known as clustered regularly interspaced short palindromic repeats (CRISPRs) within bacterial genomes (Ishino et al., 1987; Mojica et al., 2000) and subsequent gene editing in mammalian cells (Cong et al., 2013; Mali et al., 2013), CRISPR-Cas9 has become the cutting edge option for inexpensive and efficient gene editing. With successful application in cellular systems ranging from tobacco plant cells to zebrafish and primary human cells (Hsu et al., 2014), CRISPR-Cas9 can be directed by design of a short 20 nucleotide RNA sequence to create targeted DNA double strand breaks (DSB) within large genomes (Park et al., 2016). After DSBs occur, cells can initiate repair either through high fidelity homologous recombination (HR) or error-prone non-homologous end joining (NHEJ), often leading to small insertion and deletion (indel) mutations resulting in gene knockout (Gaj et al., 2013; Bétermier et al., 2014) (Figure 1).
Figure 1. Principle of genome editing by CRISPR Cas9. The principle of a gene knockout by CRISPR-Cas9 is shown exemplarily for the RIP1 sequence. A. Single guided RNA (sgRNA) consists of the target sequence specific crRNA (CRISPR RNA) and the constant tracrRNA (trans-activating crRNA) (Jinek et al., 2012). crRNA is binding to the genomic DNA adjacent to the PAM motif and tracrRNA guides the Cas9 enzyme to the locus. B. Cas9 mediated DNA double strand breaks (DSB) activate non-homologous end joining (NHEJ). C. Imprecise DSB repair leads to gain or loss of nucleotides (indels) with a two-thirds chance of causing frameshift mutations that may result in the generation of premature stop codons.
A number of different strategies have emerged to deliver Cas9 protein and targeting RNA into cells, including electroporation or transfection of Cas9/sgRNA ribonucleoprotein complexes, mRNA, plasmid or lentiviral vectors carrying sgRNA and Cas9 payloads (Sander and Joung, 2014; Shalem et al., 2014). Previously these LentiCRISPR plasmids carried a resistance gene to allow selection of cells with constitutive expression of the machinery necessary for CRISPR-Cas9-directed gene disruption.
As shown in our recent publication (McComb et al., 2016), we have adapted the LentiCRISPR protocol for directed disruption of several genes simultaneously in cell lines and primary leukemia cells based on selection by fluorescence combined with fluorescence-activated cell sorting (FACS). By swapping the puromycin resistance gene for fluorescent protein markers (EGFP, mCherry, tagBFP, or RFP657), up to four genes can be simultaneously targeted for CRISPR-Cas9-mediated gene disruption. Fluorescence-activated cell sorting enables isolation of cell lines or primary human cells bearing sgRNAs targeting one to four genes in one single experimental step. Our multi-color LentiCRISPR technique thus allows the simultaneous generation of knockout cells bearing anywhere between one and four gene knockouts, allowing rapid testing of gene-gene interaction within a set of genes of interest. The backbone vectors with the four different fluorescence markers as well as the herein described target constructs including cloning information have been deposited at Addgene.
Here we provide a complete step-by-step guide protocol to generate single and multi-gene knockout cells by multicolor LentiCRISPR (see Figure 2 for schematic overview).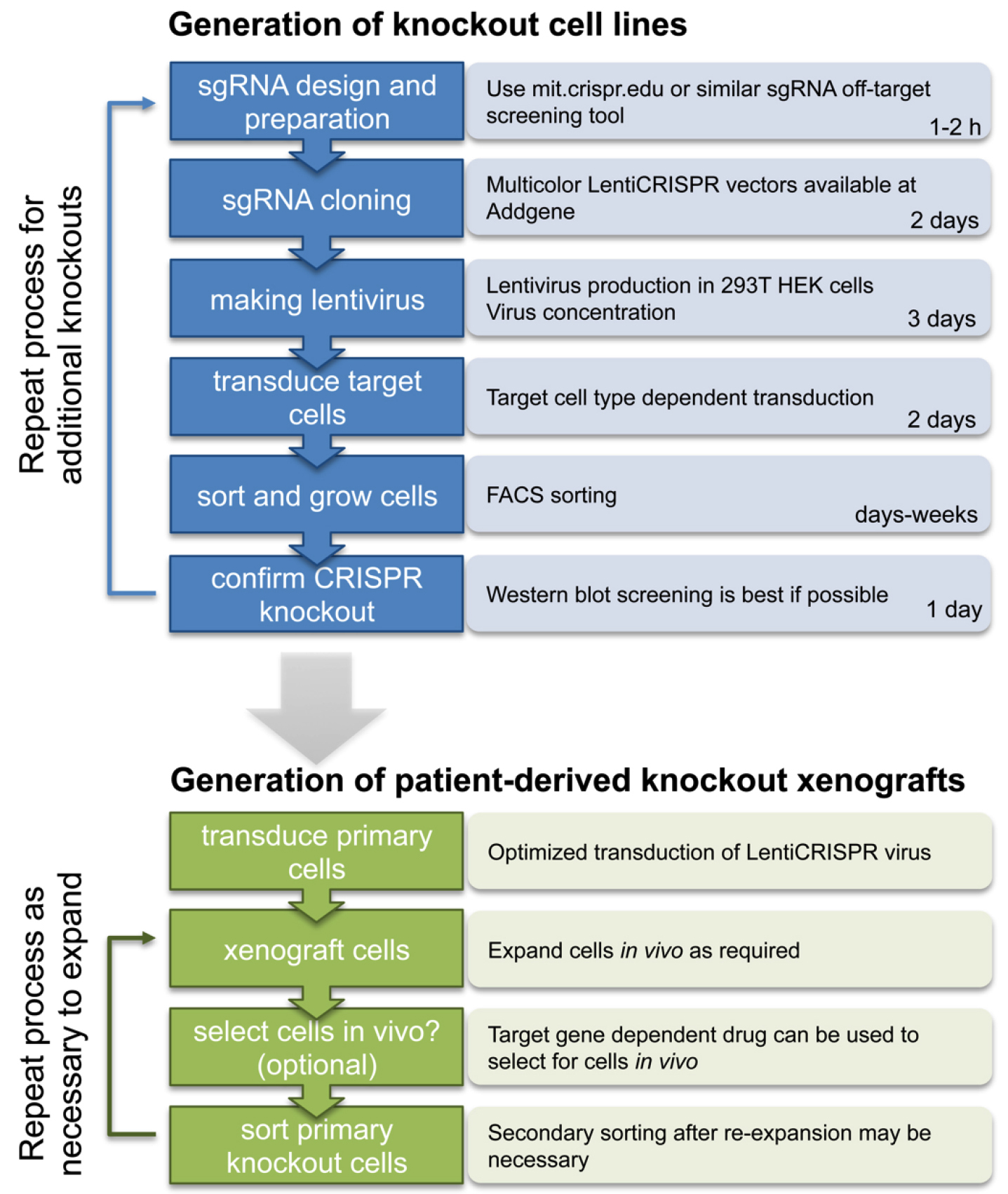
Figure 2. Schematic overview of the procedure to generate multicolor LentiCRISPR knockout cell lines and patient derived xenografts
Development of the protocol
To study the regulation of cell death in leukemia, we developed multi-color LentiCRISPR as a tool to target proteins essential for two divergent pathways of programmed cell death, apoptosis and necroptosis (McComb et al., 2016). Efficient deletion of the respective targets, like RIP1, RIP3, MLKL, FADD and CASP8, was demonstrated by Western blot analysis of protein in targeted compartments (see Data analysis). Through simultaneous gene disruption, we showed that it is necessary to inactivate both apoptosis and necroptosis within leukemic cell lines and patient-derived xenografts in order to render cells resistant to SMAC mimetics, a specific class of chemotherapeutic compounds targeting the inhibitor of apoptosis proteins, IAPs. These data provide convincing evidence that both apoptosis and necroptosis can independently kill leukemia cells in vivo, and are a strong proof of concept for the multicolor LentiCRISPR technique as a means to investigate gene redundancy.
Experimental design
sgRNA design and preparation. We first describe a fast and easy way to design and clone sgRNAs for any target gene with a single pot reaction for restriction and ligation (Figure 3). We have had good success utilizing the CRISPR design online tool (http://cripr.mit.edu) from the Massachusetts Institute of Technology, developed by the lab of Feng Zhang to predict binding sites for Cas9 with minimal risk of off-target activity (Hsu et al., 2013). Alternative sgRNA prediction software (such as http://crispor.tefor.net/) can also be used to provide in silico prediction of sgRNA-specific cleavage activity based on a number of different algorithms. However, we still recommend the design of three sgRNAs per target gene and assessing their gene knockout activity in cell lines before moving on to more challenging applications. Strategies targeting only the 5’ exon of candidate genes might induce in-frame mutations that can retain the full protein functionality. A recent publication showed that targeting particular exonic regions with key functional protein domains increases the chance of null mutations without a full protein knockout (Shi et al., 2015). For this reason, we suggest spreading sgRNA candidates among different exons to increase the probability of achieving a potent gene deletion.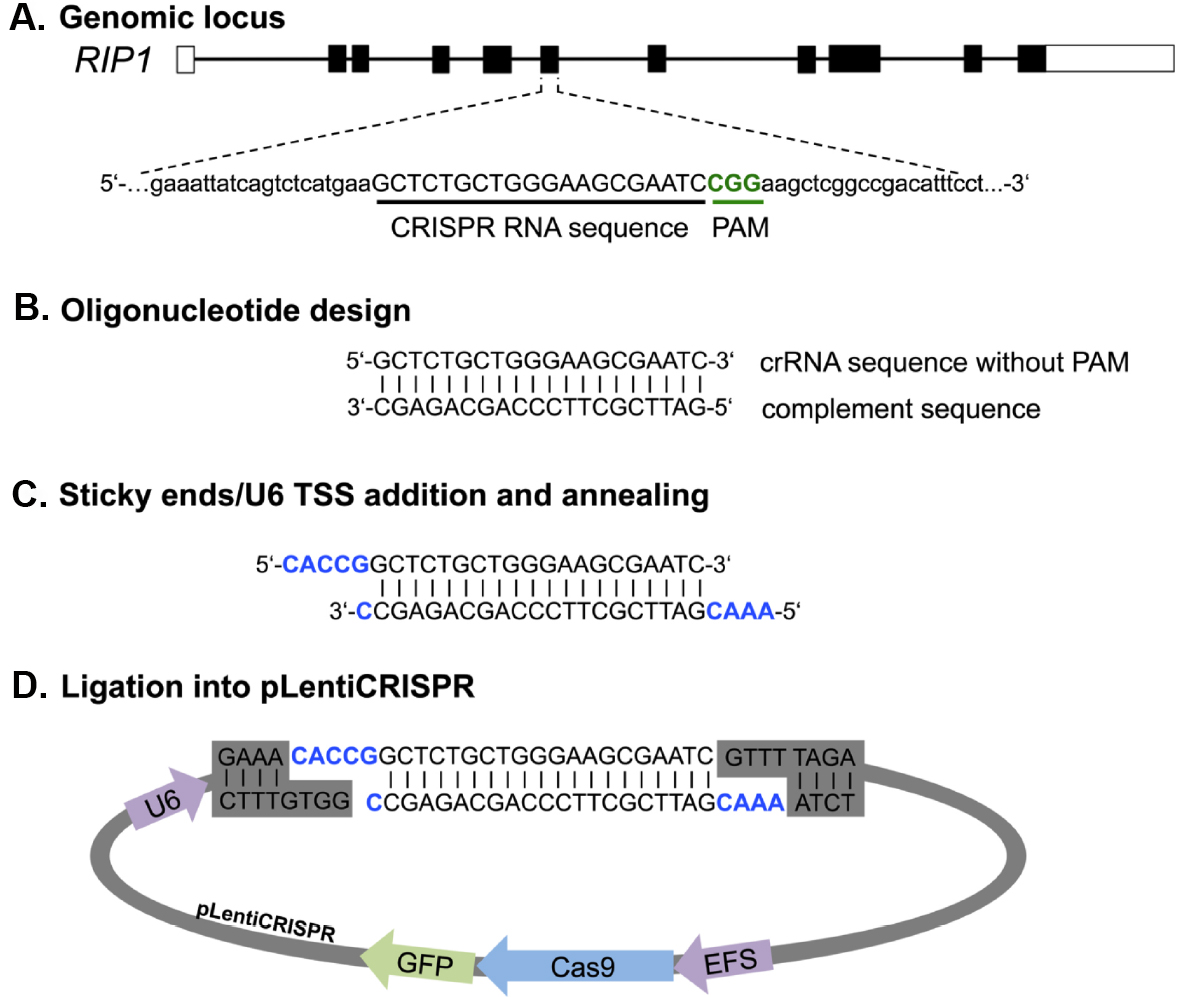
Figure 3. Principle of primer design and cloning for LentiCRISPR-Cas9 mediated gene knockout. Primer design is shown exemplarily for RIP1. A. From the genomic sequence the target locus for CRISPR editing was chosen and screened by http://crispr.mit.edu for sgRNA binding sites. B. After choosing a guide by score and location, complement primer sequence can be generated. C and D. For ligation into the Esp3I restriction site of the pLentiCRISPR plasmid sticky ends (CACC/CAAA) and the U6 transcriptional start site (TSS), (G) has to be added to the oligonucleotide sequence. U6, RNA Pol III promoter; EFS, EF1 short promoter.
Lentiviral production and infection. This protocol makes use of multicolor LentiCRISPR plasmids cloned from one-vector LentiCRISPR system developed by the Zhang lab (Shalem et al., 2014). Protocol conditions have been optimized for the transduction of acute lymphoblastic leukemia (ALL) cell lines and patient derived xenograft ALL cells from previously published protocols (Tiscornia et al., 2006; Kutner et al., 2009; Weber et al., 2012). Optimized conditions are recommended for every cell line or patient-derived sample. Here, we describe the production of lentiviral particles with the VSV-G envelope, because it is known for its high titers and broad tropism. Depending on the target cells the pseudotyping can be exchanged. For murine applications, the exchange to a mouse ecotropic envelope protein enhances safety and enables the use of the lentiviral vectors under biosafety level 1 conditions.
Analysis of knockout efficiency
After purification of cells transduced with Cas9/sgRNA targeted against a gene of interest, it is straightforward to confirm knockout at protein level (measurement via flow cytometry, ELISA or Western blot). Thus it is essential that a specific antibody for your protein of interest is available (see Figure 4 for further discussion and considerations for single cell cloning). Regardless of the viral transduction efficiency and the gene-editing efficacy of the selected sgRNA/CRISPR-Cas9 construct, DNA mutations do not invariantly lead to a loss of protein. Based on the triplet coding sequences the ratio of a frameshift after NHEJ is 2:1 resulting in possible indels without frameshift. Depending on the location of indel formation, this may also lead to unpredictable effects on mRNA or protein stability. Thus, we do not recommend knockout confirmation at DNA or RNA level by sequencing or SURVEYOR nuclease assay since this does not confirm the loss of the protein expression and function. Lesions can lead to a loss of amino acids and a change in protein functionality, but can also result in a slightly impaired protein with a near wild type function. Depending on the target gene a functional assay can be performed (i.e., enzyme activity) or cellular localization can be visualized (i.e., for nuclear receptors) but we recommend performing Western blots or ELISA to quantify protein level.
Limitations of the technique
Lentiviral vector efficiently delivers the CRISPR machinery to a wide range of cell lines and primary cells. However, as with any viral approach, there is a risk of insertional mutagenesis, although this can be minimized by transducing cells at a low multiplicity of infection (MOI) to limit the number of integration events. Constitutive expression of Cas9/sgRNA may also lead to accumulation of mutations at off-target sites, stressing the need for good sgRNA design to limit off-target binding.
Efficiency of knockouts using LentiCRISPR can vary significantly depending on the specific genes targeted, especially for targets that confer a selective advantage/disadvantage to knockout cells. It might thus be a benefit to utilize an inducible CRISPR plasmid for such applications. In this case, Cas9 or sgRNA expression would be controllable in vitro and in vivo by administration of i.e., doxycycline.
Our strategy also depends on the reliability of the detection of reporter fluorescence. There is some evidence for silencing of expression over time for fluorescent proteins of the GFP family when expressed from lentiviral vectors. The populations should therefore be continuously monitored for expression. While lentiviral delivery has been proven to be very efficient in many different cellular systems, transduction efficiency heavily depends on the size of the delivered plasmids (Canté-Barrett et al., 2016). The large size of the LentiCRISPR vector is known to lead to low viral titers, nonetheless, viral concentration and other optimizations in the protocol below have allowed us to successfully apply these vectors in hard to transduce leukemic cell lines and primary cells. 
Figure 4. Considerations for single cell cloning. We recommend single cell cloning if a clonal cell population with a constant genetic background is desired for long term experimentation. Here we present a Western blot confirmation of the knockout of RIP1 in either (A) double sorted or (B) single cell cloned Jurkat cells. Irrespective the purity of the sorted cell population, a minor RIP1 signal remains in the double sorted population, whereas in single cell clones a pure knockout can be achieved. Proteins were detected with mouse anti-RIP1 (1:1,000) and goat anti-mouse-HRP (1:5,000).
Materials and Reagents
- Filtered sterile pipette tips
10 µl (STARLAB INTERNATION, catalog number: S1121-3810 )
200 µl (STARLAB INTERNATION, catalog number: S1120-8810 )
1,000 µl (STARLAB INTERNATION, catalog number: S1126-7810 ) - TC plate
6 well (SARSTEDT, catalog number: 83.3920 )
24 well (SARSTEDT, catalog number: 83.3922 )
96 well R (SARSTEDT, catalog number: 83.3925 ) - Filters: Filtropur S
0.22 µm (SARSTEDT, catalog number: 83.1826.102 )
0.45 µm (SARSTEDT, catalog number: 83.1826 ) - Serological pipettes
5 ml (SARSTEDT, catalog number: 86.1253.001 )
10 ml (SARSTEDT, catalog number: 86.1254.001 )
25 ml (SARSTEDT, catalog number: 86.1685.001 ) - Amicon Ultra-15 centrifugal filter units (EMD Millipore, catalog number: UFC900308 )
- Tube
15 ml (SARSTEDT, catalog number: 62.554.502 )
50 ml (SARSTEDT, catalog number: 62.547.254 )
13 ml for bacteria culture (SARSTEDT, catalog number: 62.515.006 ) - SafeSeal tubes 1.5 ml (SARSTEDT, catalog number: 72.706 )
- Nitrocellulose membranes (Trans-Blot Turbo transfer pack) (Bio-Rad Laboratories, catalog number: 170-4159 )
- Disposable syringe 10 ml (CODAN, catalog number: 62.6616 )
- Injectomat syringe 50 ml (Fresenius Kabi, catalog number: 9000711 )
- Petri dishes (92 x 16 mm) for LB plates (SARSTEDT, catalog number: 82.1472.001 )
- Round-bottom tube, 5 ml (Corning, Falcon®, catalog number: 352052 )
- Round-bottom tube with cell strainer cap, 5 ml (Corning, Falcon®, catalog number: 352235 )
- TC flask T175, vent. cap (SARSTEDT, catalog number: 83.3912.002 )
- Microvette 100 LH, Lithium-Heparin (SARSTEDT, catalog number: 20.1282 )
- NSG (NOD.Cg-Prkdcscid IL2rgtm1Wjl/SzJ) mice (RRID:IMSR_ARC:NSG), age ~4-6 weeks
Caution: Institutional animal care guidelines must be followed. Here the animal experiments were approved by the veterinary office and the ethics commission of the Canton of Zurich, Switzerland. - HEK293T cells (ATCC, catalog number: CRL-3216 )
Note: Cells should be growing actively in culture with DMEM medium with 10% (v/v) FBS. Cells should be maintained between 10-90% confluence, splitting at least twice per week. - Target cells
Note: Cells should be growing in healthy culture with appropriate medium. For most ALL cell lines RPMI with 10% (v/v) FBS is suitable. Primary cells should be thawed a day before transduction.
Caution: Primary human cells have to be considered potentially hazardous. Primary derived xenografts were obtained from human ALL samples recovered from cryopreserved bone marrow aspirates. Here patients were enrolled in the ALL-BFM 2000 and ALL-BFM 2009 studies. Informed consent was given in accordance with the Declaration of Helsinki and approval was granted by the Ethics Commission of the Kanton Zurich. - One Shot TOP10 Chemically Competent E. coli (Thermo Fisher Scientific, InvitrogenTM, catalog number: C404010 or equivalent)
- CRISPR target plasmid with BsmBI (Esp3I) insert site, e.g.,
pLentiCRISPR-EGFP (Addgene, catalog number: 75159 )
RFP657 (Addgene, catalog number: 75162 )
BFP (Addgene, catalog number: 75160 )
mCherry (Addgene, catalog number: 75161 ) - psPAX2 plasmid (Addgene, catalog number: 12260 )
- p.CMV.VSV.G plasmid (Addgene, catalog number: 8454 )
Caution: It may cause an allergic skin reaction or asthma symptoms. - Oligonucleotides 100 µM, standard synthesis, desalted purification (for example sequences see Table 1)
Table 1. Sequences of oligonucleotides for pLentiCRISPR cloning
- Tango buffer (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: BY5 )
- T4 DNA ligase (New England Biolabs, catalog number: M0202M ) make aliquots of T4 DNA ligase buffer and use only freshly thawed aliquots for reaction
- Esp3I restriction enzyme (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: ER0451 )
Note: NOT NEB BsmBI, BsmBI requires 55 °C for efficient cleavage – not compatible with this protocol. - SOC medium (Sigma-Aldrich, catalog number: S1797 or equivalent)
- Ampicillin (Sigma-Aldrich, catalog number: A9518 or equivalent)
- RED Taq Ready Mix PCR Reaction mix (Sigma-Aldrich, catalog number: R2523 or equivalent)
- Mini- and Midi-plasmid Preparation Kit (QIAGEN, catalog number: 27106 or equivalent)
- Tris-acetate-EDTA buffer (TAE, 25x) (Thermo Fisher Scientific, AmbionTM, catalog number: AM9870 or equivalent)
- Polyethylenimin (PEI transfection reagent) (Sigma-Aldrich, catalog number: 408727 )
Note: You must test your preparation of PEI prior to use to establish the concentration, which is necessary to get the best transduction efficiency. We generally use ~20 µg/ml final concentration.
Caution: It is toxic if swallowed. - ProFection Mammalian Transfection System (Promega, catalog number: E1200 )
- Chloroquine diphosphate salt (Sigma-Aldrich, catalog number: C6628 or equivalent)
Caution: It is harmful if swallowed. - Calcium chloride (CaCl2)
- HBS
- Polybrene (hexadimethrine bromide) (Sigma-Aldrich, catalog number: H9268 ) stock solution 8 mg/ml in ddH2O
Caution: It is harmful if swallowed. - Dulbecco’s phosphate buffered saline (PBS) (Sigma-Aldrich, catalog number: D1408 or equivalent)
- Trypsin (0.05% in PBS) with EDTA (BioConcept, catalog number: 5-51F00-I or equivalent)
- Formalin (4% formaldehyde) (Formafix, catalog number: 01-1010 )
- Polyethylene glycol (PEG 6000) (Sigma-Aldrich, catalog number: 81253 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S7653 or equivalent)
- RetroNectin Recombinant Human Fibronectin Fragment (Takara Bio, Clontech, catalog number: T100B )
Note: Prepare Retronectin according to manufacturer’s protocol, aliquot and store at -20 °C. Retronectin can be reused up to 4 times without quality decrease. - Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A9418 or equivalent)
- Hank’s balanced salt solution (HBSS) (Thermo Fisher Scientific, catalog number: 14170088 or equivalent)
- HEPES, 1 M (Thermo Fisher Scientific, catalog number: 15630056 or equivalent)
Caution: It can cause skin irritation and serious eye irritation. - Flow cytometry antibody (PE-Cy7 anti-human CD19) (BioLegend, catalog number: 302216 , RRID:AB_314246)
- NuPage MES SDS 20x running buffer (Thermo Fisher Scientific, NovexTM, catalog number: NP0002-02 ) dilute 1:20 with ddH2O for final concentration
- Pageruler Prestained Protein ladder (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 26616 or equivalent)
- Precast gels (Criterion XT 4-12% Bis-Tris 1.0 mm gels)
18 well (Bio-Rad Laboratories, catalog number: 3450124 )
26 well (Bio-Rad Laboratories, catalog number: 3450125 ) - Nonfat-dried milk (Sigma-Aldrich, catalog number: M7409 or equivalent)
Note: Prepare 5% (w/v) working solution in 1x TBS-T. Prepare fresh and store at 4 °C for short term. - Sodium azide (NaN3) (Sigma-Aldrich, catalog number: S2002 or equivalent)
Caution: It is fatal if swallowed or in contact with skin. May cause damage to organs if swallowed. Very toxic to aquatic life. - Secondary HRP-conjugated antibody
- Western blot primary antibodies
Mouse anti-RIP1 (BD, BD Biosciences, catalog number: 551042 , RRID:AB_394015)
Rabbit anti-FADD (Cell Signaling Technology, catalog number: 2782 , RRID:AB_2100484)
Mouse anti-Tubulin (Sigma-Aldrich, catalog number: T9026 , RRID:AB_477593)
Rat anti-MLKL (EMD Millipore, catalog number: MABC604 )
Mouse anti-CASP8 (Cell Signaling Technology, catalog number: 9746 , RRID:AB_2068482)
Rabbit anti-RIP3 (Abnova, catalog number: PAB0287 , RRID:AB_1019004) - Western blot secondary antibodies conjugated to horseradish peroxidase
Goat anti-mouse (Cell Signaling Technology, catalog number: 7076 , RRID:AB_330924)
Goat anti-rabbit (Cell Signaling Technology, catalog number: 7074 , RRID:AB_10697506)
Goat anti-rat (Cell Signaling Technology, catalog number: 7077 , RRID:AB_10694715) - Baytril, Bayer, 0.5 ml in 250 ml autoclaved drinking water (stock 2.5%)
- Gene Ruler 1 kb Plus DNA ladder (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: SM1331 or equivalent)
- GelRed, 10,000x (Biotium, catalog number: 41003 )
Note: GelRed is superior to ethidium bromide by having low toxicity and high sensitivity. - Sodium hydroxide (NaOH) (Sigma-Aldrich, catalog number: 221465 or equivalent)
Caution: It is dangerous. Causes severe skin burns and eye damage. - SuperSignal® West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 34096 )
- Benzonase Nuclease (Sigma-Aldrich)
- Bacto tryptone (BD, BactoTM, catalog number: 211705 or equivalent)
- Yeast extract (BD, BactoTM, catalog number: 212750 or equivalent)
- Agar (BD, BactoTM, catalog number: 214010 or equivalent)
- Agarose (Eurogentec, catalog number: EP-0010-05 or equivalent)
- DMEM medium (Sigma-Aldrich, catalog number: D5546 or equivalent)
- Fetal calf serum (FCS) (Sigma-Aldrich, catalog number: 12133C or equivalent)
- L-glutamine, 200 mM (Thermo Fisher Scientific, GibcoTM, catalog number: 25030081 or equivalent)
- Sodium pyruvate, 100 mM (Thermo Fisher Scientific, GibcoTM, catalog number: 11360070 or equivalent)
- RPMI medium (Sigma-Aldrich, catalog number: R8758 or equivalent)
- Penicillin-streptomycin (Pen/Strep) (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 or equivalent)
- FBS
- Dimethylsulfoxide (DMSO) (Sigma-Aldrich, catalog number: D8418 or equivalent)
- Trizma HCl (Sigma-Aldrich, catalog number: T5941 or equivalent)
- Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: L3771 or equivalent)
Caution: It is harmful if swallowed and toxic in contact with skin. Flammable solid. Can cause eye and skin irritation and may cause respiratory irritation. - Bromophenol blue (Bio-Rad Laboratories, catalog number: 161-0404 or equivalent)
- Glycerol (Sigma-Aldrich, catalog number: G5516 or equivalent)
- 2-mercaptoethanol (Sigma-Aldrich, catalog number: M6250 or equivalent)
Caution: It is toxic if swallowed or if inhaled. Fatal in contact with skin. Causes skin irritations and eye damage. - Ammonium chloride (NH4Cl) (Carl Roth, catalog number: K298.1 or equivalent)
Caution: It is harmful if swallowed. It causes serious eye irritation. - Potassium chlorate (KClO3) (EMD Millipore, catalog number: 104854 or equivalent)
Caution: It is dangerous and harmful if swallowed or inhaled. May cause fire, is a strong oxidizer. - Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA) (Sigma-Aldrich, catalog number: E5134 or equivalent), prepare 0.5 M stock solution with ddH2O and pH 8.0 with NaOH
Caution: It is harmful if inhaled. - Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P9541 or equivalent)
- Trizma base (Sigma-Aldrich, catalog number: 93352 or equivalent)
- Hydrochloric acid (HCl) (Sigma-Aldrich, catalog number: H1758 or equivalent)
Caution: It is toxic if inhaled. Causes severe skin burns and eye damage. - Tween 20 (Sigma-Aldrich, catalog number: P9416 or equivalent)
- Ponceau S (Sigma-Aldrich, catalog number: P3504 )
- Acetic acid (Sigma-Aldrich, catalog number: A6283 or equivalent)
Caution: It is flammable and causes severe skin burns and eye damage. - LB agar plates Combine (see Recipes)
- LB medium Combine (see Recipes)
- DMEM complete (see Recipes)
- RPMI medium (see Recipes)
- Freezing medium (see Recipes)
- 3x SDS lysis buffer (see Recipes)
- 1x SDS loading buffer (see Recipes)
- Red blood lysis buffer (RBC buffer) (see Recipes)
- 10x TBS (see Recipes)
- 1x TBS-T (see Recipes)
- Ponceau S solution (see Recipes)
- SDS Glycine stripping buffer (see Recipes)
Equipment
- Pipettes (Gilson)
- Pipetus pipetting aid (Hirschmann Laborgeräte)
- Shaker (Thomas Scientific, model: Rocker Gyratory Lab Scale TSSL3, catalog number: 51900-27 )
- Thermocycler
- Microbiological incubator with 37 °C, atmospheric CO2 (Thermo Fisher Scientific, Heraeus)
- HeraCell 150 incubator with 37 °C, 5% CO2 (Thermo Fisher Scientific, model: HeraCell 150 incubator )
- MUPID-One electrophoresis system (LABGENE Scientific, model: MUPID-One electrophoresis system )
- Bench-top centrifuge (Eppendorf, model: 5417 C )
- Nalgene Oak Ridge centrifuge tube (Lab Depot, catalog number: 21009-386-PK )
Note: These tubes are appropriate for ultracentrifugation and can’t be exchanged by standard tubes. - Water bath (Thermo Fisher Scientific)
- Thermomixer comfort (Eppendorf)
- Vortex mixer Vortex-Genie 2 (Scientific Industries, model: Vortex-Genie 2 )
- FACSARIA III flow cytometry sorter (BD, model: FACSAria III )
- FACS Canto II flow cytometer (BD, BD Biosciences, model: FACSCANTO II )
- Gel documentation (Syngene)
- Heraeus Multifuge 3S (Thermo Fisher Scientific, model: HeraeusTM MultifugeTM 3S )
- Laminar flow hood for sterile tissue culture, biosafety level 2 approved (Thermo Fisher Scientific)
- Mastercycler nexus (Eppendorf)
- Mr. Frosty freezing container (Thermo Fisher Scientific)
- Neubauer chamber (BRAND)
- TransBlot Turbo transfer system (Bio-Rad Laboratories, model: Trans-Blot® TurboTM transfer system )
- Western blot chamber (Criterion Cell) and PowerPac basic power supply (Bio-Rad Laboratories)
- Spatula
Software
- A Plasmid Editor (http://biologylabs.utah.edu/jorgensen/wayned/ape/) (Optional)
- Oligonucleotide Properties Calculator (http://biotools.nubic.northwestern.edu) (Optional)
Procedure
Part I. sgRNA design and preparation
- Design of sgRNA sequence (Timing 1-2 h)
- Download genomic sequence from genome database (e.g., NCBI in FASTA format).
- Open your sequence with ‘A plasmid Editor’ (http://biologylabs.utah.edu/jorgensen/wayned/ape/) or similar sequence analysis software.
- Choose a target locus for CRISPR editing. Due to the variability of CRISPR knockout efficiency we generally choose 3 different exonic regions if possible. See introduction for discussion of additional considerations for target locus selection.
- Screen your target genomic locus for CRISPR guide RNA binding sites by inputting specific sequences into an online tool such as http://crispr.mit.edu.
Note: Initial work was done with the crispr.mit.edu online tool. But alternative CRIPSPR calculators like http://crispor.tefor.net/ may offer additional prediction efficiency. - Choose guides by score and location in the genome to determine targeting sites. Cas9 will cut 3 bp upstream of PAM site. We recommend choosing three different sgRNAs to improve the probability of achieving a full knockout.
- Copy the 20 nucleotide sequence of the selected guide RNA(s) without PAM and generate the complement sequence by using a bioinformatics software tool such as e.g., the Oligonucleotide Properties Calculator (http://biotools.nubic.northwestern.edu).
- For ligation into pLentiCRISPR plasmids add additional nucleotide to the sequence to generate sticky ends complement to the Esp3I restriction site (CACC/AAAC) and the transcriptional start site for RNA polymerase III (single G/C).
Oligonucleotide Additional sequence (5’-3’) Fw_sg CACCG(N20) Rev_sg AAAC(N20)C - Order the forward and reverse oligonucleotides (simple desalted oligonucleotides are adequate for this protocol).
- See example oligonucleotide design strategy for RIP1 in Figure 3.
- Download genomic sequence from genome database (e.g., NCBI in FASTA format).
- Annealing of single-stranded oligonucleotides (Timing 2.5 h)
- For lyophilized oligonucleotides, add an appropriate volume of ddH2O to obtain stock concentration of 100 µM.
- Pipette the following reaction for each pair of forward and reverse oligonucleotide.
Component Amount Fw_sg oligo 10 µl Rev_sg oligo 10 µl Tango buffer 10 µl ddH2O 70 µl - Bring up to 95 °C for 5 min in a thermocycler.
- Anneal the oligonucleotides by cooling to room temperature over 1-2 h.
- For lyophilized oligonucleotides, add an appropriate volume of ddH2O to obtain stock concentration of 100 µM.
- Ligation of double-stranded oligonucleotides into pLentiCRISPR (Timing 6 h)
- Set up following single pot reaction for plasmid restriction and ligation of annealed double-stranded oligonucleotides into pLentiCRISPR.
Component Amount pLentiCRISPR 150 ng double-stranded oligos (from step B4, Part I) 1 µl NEB T4 ligase buffer (10x) 2 µl ddH2O up to 18 µl Esp3I 0.5 µl T4 DNA ligase 0.5 µl - Run the following reaction in a thermocycler.
Note: There is an additional BsmBI (Esp3I) site in the tagBFP sequence in the pLentiCRISPR-BFP vector. For this vector the final 37 °C step should be removed from the cycle protocol to improve yield.Cycle Temperature Time Ligation (15 cycles) 37 °C 5 min 16 °C 10 min Final 37 °C 15 min 80 °C 5 min Hold 4 °C
PAUSE POINT: After heat inactivation of Esp3I the plasmid can be immediately transformed into bacteria or stored at -20 °C.
- Set up following single pot reaction for plasmid restriction and ligation of annealed double-stranded oligonucleotides into pLentiCRISPR.
- Transformation and identification of bacterial clones with pLentiCRISPR and double-stranded oligonucleotide insert (Timing 3 d)
Day 1:- Transform ligation mix into competent bacteria.
- Add 2 µl ligation mix into ice-cold competent bacteria (50 µl).
Note: Do not use more than 5 µl ligation product for transformation of 50 µl competent bacteria. - Incubate mixture on ice for 20 min and heat-shock it at 42 °C for 90 sec.
- Cool down on ice for 2 min.
- Add 200 µl SOC medium (without antibiotics) and incubate for 45 min at 37 °C with gentle shaking.
- Plate 100 µl bacteria suspension on LB plate containing 100 µg/ml ampicillin.
- Incubate overnight at 37 °C in the microbiological incubator.
Day 2:
- Run a colony PCR for 3-5 colonies per transformed pLentiCRISPR.
Note: No colonies? See notes in Table 2. - Prepare separate LB-agar plate with a number for each clone that will be picked.
- Prepare PCR reaction mix. Use Fw_U6 as forward primer and the sequence specific Rev_sg oligonucleotide as reverse primer.
Component Amount RED Taq Ready mix (2x) 5 µl ddH2O 4 µl Fw_U6 (10 µM) 0.5 µl Rev_sg oligo (10 µM) 0.5 µl - Pick single colonies with pipette tip, streak on separate numbered plate and subsequently submerge pipette tip in one well of PCR reaction buffer.
- After picking all colonies, remove pipette tips, run PCR and put numbered plate with streaks at 37 °C for 2-5 h in the microbiological incubator.
- Colony PCR program:
Cycle Temperature Time Initial 95 °C 3 min Cycle (25 cycles) 95 °C 30 sec 55 °C 30 sec 72 °C 10 sec Final 72 °C 10 min Hold 4 °C - Load PCR reactions on an agarose gel (2% [w/v] agarose/TAE buffer) (see Figure 5 for estimated product).
Note: No positive colonies? Too many colonies? See notes in Table 2.
Figure 5. Example of colony PCR products. pLentiCRISPR clones with inserted double-stranded oligonucleotides give a band around 100 bp. Empty vectors should give no band and will show only the primer dimers. - Pick positive clone(s) from numbered plates to start overnight cultures in LB medium at 37 °C in the microbiological incubator.
Day 3:
- Isolate plasmid DNA by mini or midi prep according to manufacturer’s protocol.
PAUSE POINT: Plasmids can be immediately transfected into HEK293T cells or stored at 4 °C or -20 °C.
- Transform ligation mix into competent bacteria.
Part II. Lentiviral production and infection
- Production of pLentiCRISPR lentiviral particles (Timing 3-5 d)
Day 1:- In the afternoon seed 500,000 HEK293T cells per 6 well plate in 2 ml of DMEM complete.
Note: Keep HEK293T cells always subconfluent. We also recommend working without antibiotics as we generally achieve higher titers without antibiotics in the medium. This protocol details the production of pilot-scale batches of viral supernatant (approximately 6-9 ml) appropriate for cell line applications but can be scaled up using T175 flasks to produce larger batches. - Incubate overnight to allow HEK293T cells to adhere and equilibrate.
Day 2
- Transfect HEK293T cells with lentiviral plasmids (pLentiCRISPR, psPAX2 and pVSV.G). We recommend using one of the two following techniques.
Notes:
a.Be careful not to disturb the cells while you add the transfection reagents.
b.We use a ratio of 8:3:3 for LentiCRISPR:psPAX2:pVSV.G resulting in 4 µg pLentiCRISPR, and 1.5 µg of each of psPAX2 and p.CMV.VSV.G per well. But plasmid ratio should be titrated. - PEI transfection reagent
- Thaw all reagents to room temperature (RT, about 22 °C).
- Dilute the plasmids in 1 ml of serum free, antibiotic free DMEM.
- Add 20 µl of PEI reagent and vortex immediately.
- Incubate the PEI/plasmid mixture for exactly 20 min at RT.
- Add the mixture dropwise directly to the HEK293T cells.
- ProFection mammalian transfection system
- Thaw all reagents to RT.
- Replace the cell culture medium with 3 ml fresh antibiotic free DMEM complete and add 3 µl chloroquine (final concentration 25 µM).
- Mix plasmids with 2 M CaCl2 and add ddH2O up to 150 µl.
- Gently add DNA-CaCl2 solution dropwise to 150 µl HBS while vortexing.
- Incubate for 30 min at room temperature.
- Add the mixture dropwise directly to the HEK293T cells.
Note: All subsequent steps are subject to biosafety level 2 regulations. Perform all work in a biosafety cabinet and do not use vacuum to suction supernatants to avoid aerosolizing virus.
- After 4-6 h, remove the transfection medium and carefully replace by 3 ml pre-warmed DMEM complete. For PEI transfection, medium change is not strictly necessary, no obvious toxicity to cells could be observed.
Day 3:
- The next morning examine the transfection efficiency by fluorescence microscopy. Some fluorescent signal should be visible in the majority of cells, although signal can be expected to increase for up to 72 h.
Note: No fluorescence? See notes in Table 2. - Collect the supernatant from the HEK293T cells and replace it with 3 ml pre-warmed DMEM complete medium. Keep viral supernatant on ice.
- Put HEK293T cells back to incubator (5% CO2) until next day.
- Filter the supernatant through a 0.45 µm filter to clear any remaining cells and freeze an aliquot of 100 µl for titer test at -80 °C.
- Supernatant can be kept at 4 °C in order to combine supernatant collected on subsequent days.
Note: Do not user smaller filter size as it will decrease virus titer.
Day 4:
- Repeat steps A6-A9 (Part II).
Day 5:
- Repeat steps A6-A9 (Part II).
Note: You can harvest viral supernatant up to 72 h after transfection, but the viral titer is decreasing after 48 h. Control the appearance of the HEK293T cells, cells should not overgrow massively. The highest titer is normally reached 24 h after transfection.
PAUSE POINT: Supernatant can be immediately concentrated (see step C1, Part II) or aliquoted and stored at 4 °C for up to 1 week, or at -80 °C with a marginal decrease in viral titer (> 1 year). Freeze and thaw cycles should be avoided, as it will reduce titer.
- In the afternoon seed 500,000 HEK293T cells per 6 well plate in 2 ml of DMEM complete.
- Titration of pLentiCRISPR lentiviral particles (Timing 3 d)
Note: For initial screening of sgRNA functionality viral supernatants can be used directly on target cells without titration, however it is recommended to still perform titrations as viral aliquots can be stored and used for later experiments.
Day 1:- Plate 50,000 HEK293T cells in 0.5 ml DMEM complete per well in a 24 well plate.
- Work in triplicates.
Note: HEK293T titers are a relative measurement of infectious units, thus we recommend working always with HEK293T cells under the same conditions (incubation times, volumes etc.). Stable processes enables to compare the titers between different productions. - Add 0.5 µl polybrene (final concentration of 8 µg/ml).
- Incubate HEK293T cells for 6 h in the incubator (5% CO2).
- Add viral supernatant in increasing amounts to the medium (20 µl-100 µl of unconcentrated virus).
CAUTION: All subsequent steps are subject to biosafety level 2 regulations. Do not use the vacuum pump to suction supernatants to avoid aerosolizing virus.
Note: If this is done for the first time use a broad range of concentrations (1/10 to 1/100,000 relative dilutions of viral supernatant) to ensure that the data will be evaluable.- Centrifuge the 24 well plate for 1 h, 750 x g, RT and keep overnight in the incubator (5% CO2).
Day 2:
- Replace medium by 0.5 ml fresh DMEM complete and incubate for 48 h.
Day 3:
- Remove supernatant and wash cells with PBS.
- Add 0.5 ml trypsin/EDTA and incubate for 5 min at 37 °C until the cells detach from the surface.
- Add 0.5 ml DMEM complete, resuspend the cells and transfer into FACS tubes.
- Spin for 5 min, 750 x g, RT.
- Remove supernatant and resuspend the cells in 0.5 ml PBS with 2% (v/v) formalin.
- Measure the transduction efficiency via flow cytometry.
- The linear range of viral transduction is in between 5-30%. Calculate the titer only for those wells that are comprised. The titer is calculated with the following formula. For more details refer to Weber et al. (2012).
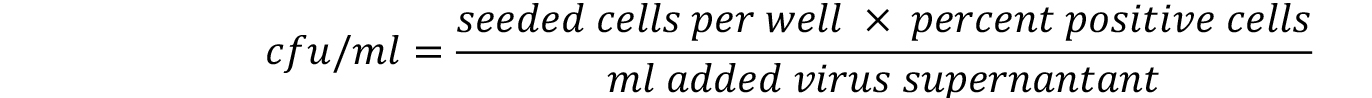
Notes: - Under the procedure described in this protocol we generally expect to achieve viral supernatants that show cfu/ml above 1 x 105.
- Low titer? See notes in Table 2.
- Plate 50,000 HEK293T cells in 0.5 ml DMEM complete per well in a 24 well plate.
- Concentration of lentiviral particles (Timing 1-8 h)
- To increase the titer and therefore the transduction potential you can concentrate the virus by (a) column, (b) ultracentrifugation or (c) PEG 6000 precipitation.
- Concentration by Amicon centrifugal filter units
- Centrifugation speed and time should be optimized.
- We recommend loading 15 ml viral supernatant into the upper chamber and centrifuge for 10 min at 1,800 x g at 4 °C.
- Volume of viral supernatant should be decreased to 1 ml.
- Concentration index 1:15.
- Concentration by ultracentrifugation
- Load up to 40 ml viral supernatant to a Nalgene Oak Ridge centrifuge tube.
- Centrifuge for 6 h, at 8,000 x g, 4 °C.
- Afterwards virus is visible as a transparent jelly pellet.
- Discard supernatant carefully and resuspend pellet by vigorously pipetting in 1 ml medium of your choice.
- Concentration index: 1:40.
- Concentration by PEG 6000 precipitation
- Day 1. To 5 ml virus supernatant, add 1.3 ml of 50% PEG 6000 solution, 0.54 ml 4 M NaCl and 0.59 ml PBS. The final PEG 6000 concentration will be 8.5% and the final NaCl concentration will be ~0.3 M.
- Mix contents and store at 4 °C overnight.
- Day 2. Centrifuge mixture for 30 min at 2,000 x g, 4 °C.
- After centrifugation a white virus pellet is visible.
- Carefully decant the supernatant and resuspend the pellet in 100-500 µl medium by vigorously pipetting liquid up and down.
- Concentration index 1:50.
- Transduction of cell lines with pLentiCRISPR lentiviral particles (Timing 4 d)
Note: This protocol is optimized for human leukemia cell lines (e.g., Jurkat). See below for optimized protocol for primary leukemic cells (step E1, Part II).
CAUTION: All subsequent steps are subject to biosafety level 2 regulations. Do not use the vacuum pump to suction supernatants to avoid aerosolizing virus.
Day 1:- Count target cells and place 1 million cells in 150 µl RPMI medium per well in a 24 well plate.
- Add polybrene to the target cells at a concentration of 8 µg/ml.
- Thaw viral supernatant quickly in a water bath and keep it on ice as soon as it is melted.
- Add 50 µl-1 ml viral supernatant(s) on top of the cells, depending on desired MOI (multiplicity of infection).
Note: For multiple target knockouts mix virus particles to transduce with the mixture in one step or perform serial transduction. In particular for triple and quadruple knockouts serial transduction can improve efficiencies. - Gently shake the plate to mix the cells with the viral supernatant.
- After 2 h bring up to 2 ml with fresh RPMI medium.
- Incubate overnight at 37 °C.
Day 2:
- Centrifuge the cells and gently remove the supernatant by pipetting and replace with 1 ml fresh RPMI.
Day 3:
- Repeat steps D1-D8 (Part II). You can repeat viral transduction up to three times if higher transduction rates are desired.
Day 4:
- Take a small aliquot of cells and fix by adding the same volume of 4% formalin (final concentration 2%) and incubate on ice for 10 min. Proceed to determine the transduction efficiency via flow cytometry.
- For a pure knockout cell line we recommend performing single cell clones either by limiting dilution or flow cytometry sort. But for a first validation of knockout efficiency and biological phenotype of the knockout we recommend using the bulk population after one or two rounds of flow cytometry sorting.
Notes:- Cells should not be transduced with more than 70-80% efficiency because this leads to multiple insertion, toxicity and higher risk of insertional mutagenesis.
- Low transduction efficiency? Too high transduction? See notes in Table 2.
- Cells should not be transduced with more than 70-80% efficiency because this leads to multiple insertion, toxicity and higher risk of insertional mutagenesis.
- Count target cells and place 1 million cells in 150 µl RPMI medium per well in a 24 well plate.
- Transduction of patient derived xenograft cells with pLentiCRISPR lentiviral particles (Timing 4 d)
Notes:- This protocol is optimized for human patient derived xenograft ALL cells (PDX). For some PDX it might be sufficient to use the transduction protocol with polybrene.
- Work with PDX is regulated by the Declaration of Helsinki. You should seek approval for animal experiments from your local animal care authority.
- Thaw frozen PDX cells quickly in the water bath and transfer to a 15 ml Falcon tube as soon as the suspension is solvent.
- Add 10 times volume of cold RPMI medium drop by drop to thawed cells suspension, gently shake the tube when adding.
- Wash cells by centrifugation and resuspension in fresh medium.
- Count and seed 1 million cells per well in a 24 well plate in 1 ml medium and incubate at 37 °C overnight.
Note: Low viability? See notes in Table 2. - Dilute RetroNectin to a final concentration of 0.05 mg/ml in PBS. RetroNectin can be aliquoted and frozen at -20 °C. Solution can be reused for 5 times. Thaw solution quickly in the water bath and put on ice afterwards.
- Coat a 6 well plate with 2 ml RetroNectin solution for either 2 h at room temperature or at 4 °C overnight.
Day 2:
- Remove the RetroNectin solution and block for 30 min with 2 ml 2% (v/v) BSA/PBS at room temperature. You can transfer the RetroNectin to a fresh plate to coat for the second transduction round.
- Wash the plate with 3 ml HBSS/2.5% (v/v) HEPES per well.
- Thaw viral supernatant quickly in the water bath and keep it on ice as soon as it is melted.
Note: For multiple target knockouts mix viral particles to transduce with the mixture in one step or perform serial transduction/transplantation cycles. In particular for triple and quadruple knockouts serial transduction can improve efficiencies. - Discard the washing buffer from the plate and add 2 ml viral supernatant.
- Centrifuge the plate for 20 min at 4 °C and 750 x g.
- Discard the viral supernatant but do not allow the plate to dry.
- Repeat steps E10-E12 (Part II) for 3 times.
- Transfer target cells to virus coated plate and centrifuge for 2 min at 4 °C and 750 x g.
- Incubate at 37 °C overnight.
Day 3:
- Wash the cells 3 times with 10 ml of PBS by centrifugation for 5 min at 750 x g, RT.
- Transplant the cells directly into immunocompromised mice.
- An aliquot of primary PDX cells can also be transferred onto mesenchymal stroma feeder cells to maintain their survival in vitro for later analysis.
Day 4:
- 48 h after transduction cells can be tested by flow cytometry for fluorescent protein expression to determine the transduction efficiency.
Note: Fluorescence maximum might be reached between Day 3 and Day 5, but the life span of primary cells in vitro can be shorter. We recommend transplanting cells before the transduction efficiency is directly measured via flow cytometry.
- This protocol is optimized for human patient derived xenograft ALL cells (PDX). For some PDX it might be sufficient to use the transduction protocol with polybrene.
Part III. Expansion and confirmation of knockout clones
- Expansion of knockout clones in NSG mice (Timing transplantation 2 h, engraftment weeks to months)
- Count the cells and spin down the required number. Good transplantation rates can be obtained with 1 million cells, but as few as 100 cells can successfully engraft.
- Resuspend cells in 100 µl PBS per injection.
- Transplant cells via intravenous or intrafemural transplantation (for more details see Schmitz et al., 2011).
Note: Institutional animal care guidelines must be followed. Animal experiments performed in the development of this protocol were approved by the veterinary office and the ethics commission of the Canton of Zurich, Switzerland. - Keep mice under antibiotic treatment until termination of the experiment.
- Follow up the engraftment by regular bleeding, according to local legal regulations. We use approximately 20 µl of blood taken via tail vein puncture and a Microvette tube with lithium heparin.
- Dilute blood with PBS to 100 µl and add 900 µl red blood cell lysis buffer.
- Incubate for 5 min at 4 °C.
- Centrifuge at 750 x g for 5 min at 4 °C.
- Wash cell pellet with 500 µl PBS and centrifuge again.
- Resuspend in 100 µl PBS with anti-human CD19-PECy7 antibody, as it will not interfere with any of the fluorescent proteins used here (dilution 1:1,000).
- Incubate for 20-30 min at 4 °C.
- Add 500 µl PBS and centrifuge again.
- Resuspend in 150 µl PBS and analyse by flow cytometry.
- Calculate the engraftment with following formula.
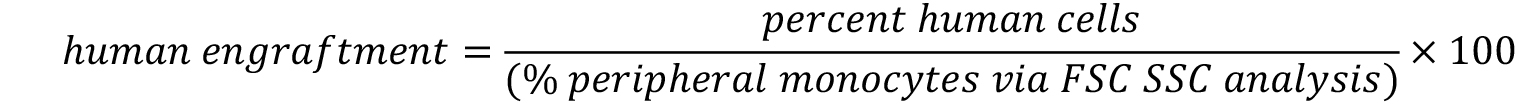
CAUTION: Criteria for euthanasia have to be applied according to the animal experimentation approval as approved by the responsible authorities.
Notes:- Depending on the PDX human engraftment can be detected in the peripheral blood already after a few days, but spleen is small. Other patients show high engraftment in the bone marrow and spleen but only low levels in the blood. For each PDX we recommend checking the blood regularly and to palpate the spleen.
- No engraftment? See notes in Table 2.
- Depending on the PDX human engraftment can be detected in the peripheral blood already after a few days, but spleen is small. Other patients show high engraftment in the bone marrow and spleen but only low levels in the blood. For each PDX we recommend checking the blood regularly and to palpate the spleen.
- To generate pure patient-derived knockout ALL cells, we suggest to do serial transplantations after sort amplification.
- To harvest the PDX cells from the spleen, sacrifice the mouse according to institutional standards and isolate the spleen from the abdomen.
- Transfer the tissue to a cell strainer, force spleen through the strainer and wash with 10 ml PBS to create a single cell suspension.
- Dilute cell suspension 1:1 with RBC buffer and incubate for 5 min at 4 °C.
- Centrifuge at 750 x g for 5 min at 4 °C and resuspend pellet in PBS.
- Count the cells and prepare for flow cytometry sort.
PAUSE POINT: PDX cells can be stored at -80 °C (short term) or in liquid nitrogen (long term). For this spin down the cells and resuspend up to 50 million cells in freezing medium and freeze carefully in a freezing container.
- Count the cells and spin down the required number. Good transplantation rates can be obtained with 1 million cells, but as few as 100 cells can successfully engraft.
- Flow cytometry sorting of single and multi-knockout cells (Timing 2 h)
- Pellet cells by centrifugation at 750 x g for 5 min at RT and resuspend at 5 million cells per ml in PBS, or a cell concentration recommended by flow facility.
- Filter cell suspension through cell strainer cap of a round-bottom tube.
- Use hierarchical gating to sort populations of single, double, triple, and/or quadruple positive cells (see below Data analysis).
- In case of low transduction efficiencies follow a serial sorting strategy to first enrich single or double positive populations with an inclusive gating strategy. Cells can then be regrown and later sorted to isolate multi-knockout cells (Figure 6).
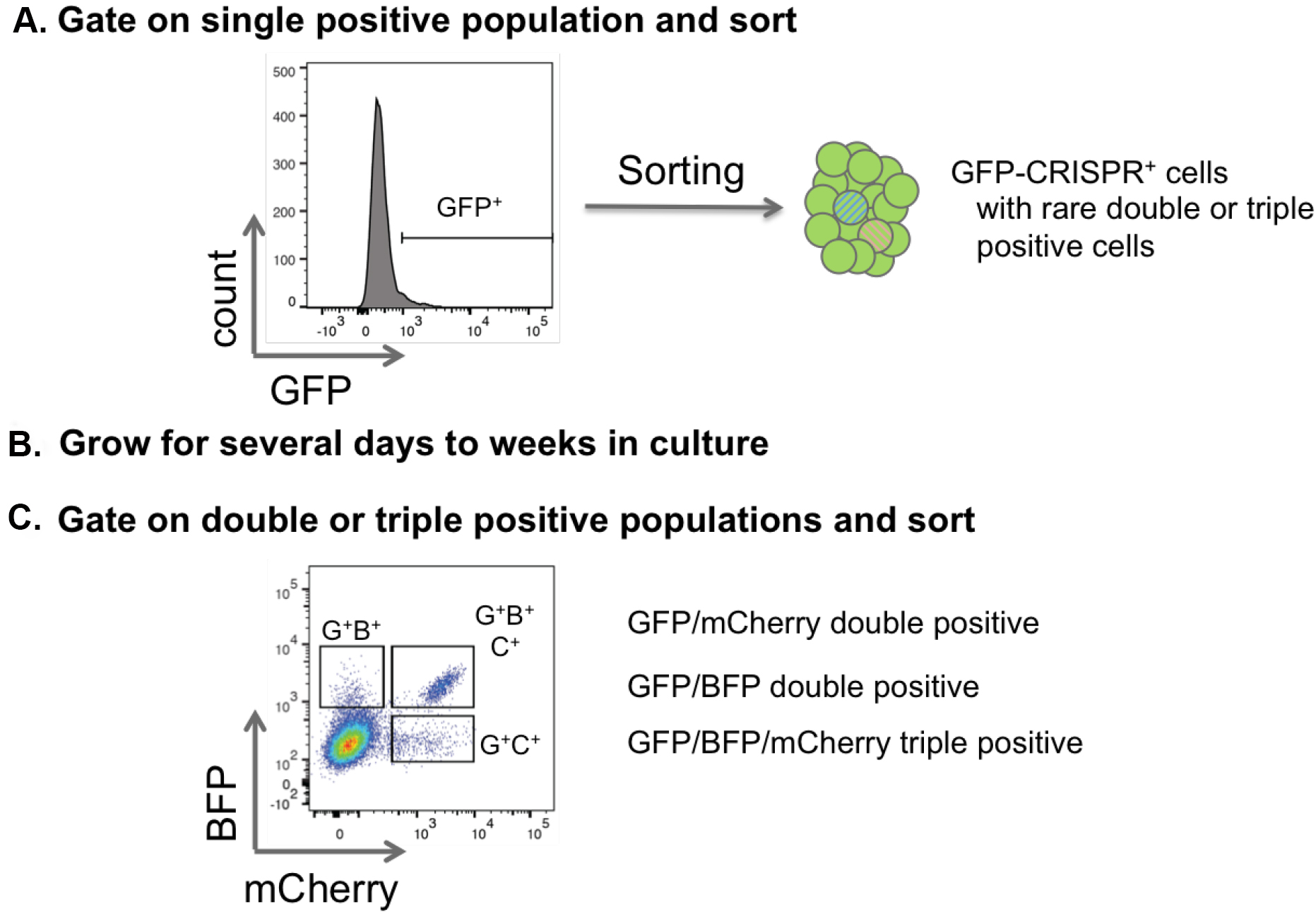
Figure 6. Sorting strategy for double and triple positive CRISPR cells. A. To purify single, double and triple transduced cells that are represented with a low frequency, first sort for one color. This population will include single, double and triple positive cells. B and C. After expansion in vitro in cell culture or in vivo in the mouse, cells can be resorted by flow cytometry specifically for the double and triple positive populations. - Return cells to culture conditions, or re-transplant into mice for later resorting and/or confirmation of knockout.
- For cell lines we recommend sorting single cell clones.
- For this prepare a 96 well plate with 100 µl medium in each well and sort one cell per well.
- Culture the cells until cell numbers are high enough to confirm the knockout by Western blot. Note that we have observed better knockout frequencies in sorted CRISPR containing cells after at least 3 weeks (and as long as 6 weeks) in culture, although this likely varies between different cell types.
- Pellet cells by centrifugation at 750 x g for 5 min at RT and resuspend at 5 million cells per ml in PBS, or a cell concentration recommended by flow facility.
- Confirmation of gene knockout by Western blot (Timing 2 d)
Day 1:- Count the cells and collect them in a 1.5 ml tube. Centrifuge at 750 x g for 5 min at RT and wash the pellet with PBS.
- Pellet the cells again and add 1x SDS loading buffer at a concentration of 300,000 cells in 80 µl.
Note: It is not recommended lysing less than 100,000 or more than 1 million cells. - Completely lyse cells by vortexing and denature the proteins by incubating the samples at 95 °C for 5 min in a thermomixer.
PAUSE POINT: After lysis samples can be stored at -20 °C for up to several months before later analysis. - Assemble a Western blot chamber with a precast gel according to the manufacturer’s instructions and remove the comb.
- Pour fresh 1x MES running buffer to the top posterior chamber and fill it completely. Pour 1x MES buffer to the front chamber until the mark or until the lower end of the comb.
Note: The 1x MES running buffer used in the top chamber must be fresh. For the front chamber the buffer can be reused for up to 3 times. - Wash the wells to remove excess of storage buffer and loose pieces of gel by pipetting up and down the running buffer inside each well with a volume of around 100 µl.
- Load the cell lysates on the gel.
- For a 26 well gel load 10-15 µl of lysate per well and 3 µl of protein ladder.
- For an 18 well gel load 20-30 µl of lysate per well and 5 µl of protein ladder.
- Load the same volume of 1x SDS loading buffer in the empty wells to make the samples run straighter.
- Cover the Western blot chamber and run the gel at a constant voltage of 120 to 140 V (it will take 1 h 30 min to 45 min approximately) until the blue mark of the samples reaches the end of the gel.
- Remove the gel from the chamber and open the plastic covers carefully with a spatula.
- Remove the top part including the wells until over the top band of the marker. Assemble the transfer stack according to manufacturer’s instructions.
- Transfer the proteins from the gel to the membrane. We routinely use a Bio-Rad protocol optimized for high molecular weight proteins: 2.5 A and 25 V for 10 min.
Note: Avoid touching the membrane with your hands. - To check that the loading was even and the transfer was complete, incubate the membrane with a Ponceau S solution for 1 min and wash it with tap water.
- Wash the membrane once with TBS-T until most of the Ponceau S stain is removed and block the membrane for 1 h with 5% milk in TBS-T.
- Incubate the membrane with the primary antibody diluted according to manufacturer’s instructions under agitation either overnight at 4 °C or for 2 h at RT. You can add sodium azide (0.01%, v/v) to store it at 4 °C and reuse it.
- Wash the membrane with TBS-T 4 x for 5 min each with agitation at RT.
- Incubate with the appropriate secondary HRP-conjugated antibody diluted 1:5,000 in 5% milk in TBS-T for 1 h at RT with agitation.
Day 2:
- Wash the membrane with TBS-T 4 x for 5 min each with agitation at RT.
- To acquire the images, cover the membrane with super signal and incubate it for 1 min. Remove excess super signal and place the membrane on a plastic cover. Acquire the chemiluminescence and the white light for the ladder without moving the membrane.
- To probe for other proteins in the same membrane, wash it once with TBST-T and strip it with SDS glycine solution for 45 min to 1 h under agitation at RT. Wash it 4 x for 5 min with TBS-T and repeat steps C14-C19 (Part III).
Note: No protein signal in positive control? No knockout? See notes in Table 2.
- Count the cells and collect them in a 1.5 ml tube. Centrifuge at 750 x g for 5 min at RT and wash the pellet with PBS.
Data analysis
The protocol described herein details an efficient method to knockout target genes in a time and cost efficient manner. We have successfully used the pLentiCRISPR Cas9 system to generate up to four knockouts simultaneously. First, a quick screen should be performed to select a sgRNA targeting sequence with good activity for generating a knockout (see introduction for considerations in sgRNA design). Here we have selected three different sgRNAs targeting the RIP1 gene, which were tested in an easily transducable cell line (Figure 7A). Sorting the fluorescent cells by flow cytometry with a subsequent Western blot will show which sgRNA presents the strongest knockout potential (Figure 7B). Here, the knockout inside of exon 6 was more efficient than knockouts in exon 9. Based on this result, a candidate sgRNA can then be used for knockout with target cell lines or PDX material. By freezing viral aliquots at -80 °C, those vectors with the highest activity can be immediately applied after initial selection of a sgRNA sequence with good activity. Four different pLentiCRISPR viral particles can be mixed and matched to enable the knockout of up to four target genes simultaneously (Figure 8). In our example, we were able to show that certain double knockout cells are resistant to treatment with SMAC mimetic birinapant in vivo (for more details see McComb et al., 2016). Figures 8A and 8B shows the distribution of single, double, triple and quadruple knockouts before (A) and after (B) selection with chemotherapeutic drug in vivo. Populations of knockout cells were isolated by fluorescence activated cell sort and retransplanted into NSG mice. After expansion in vivo cells were harvested and lysed for Western blot analysis. Here the knockout of up to four targeted genes in primary leukemia xenografts could be confirmed (Figure 8C).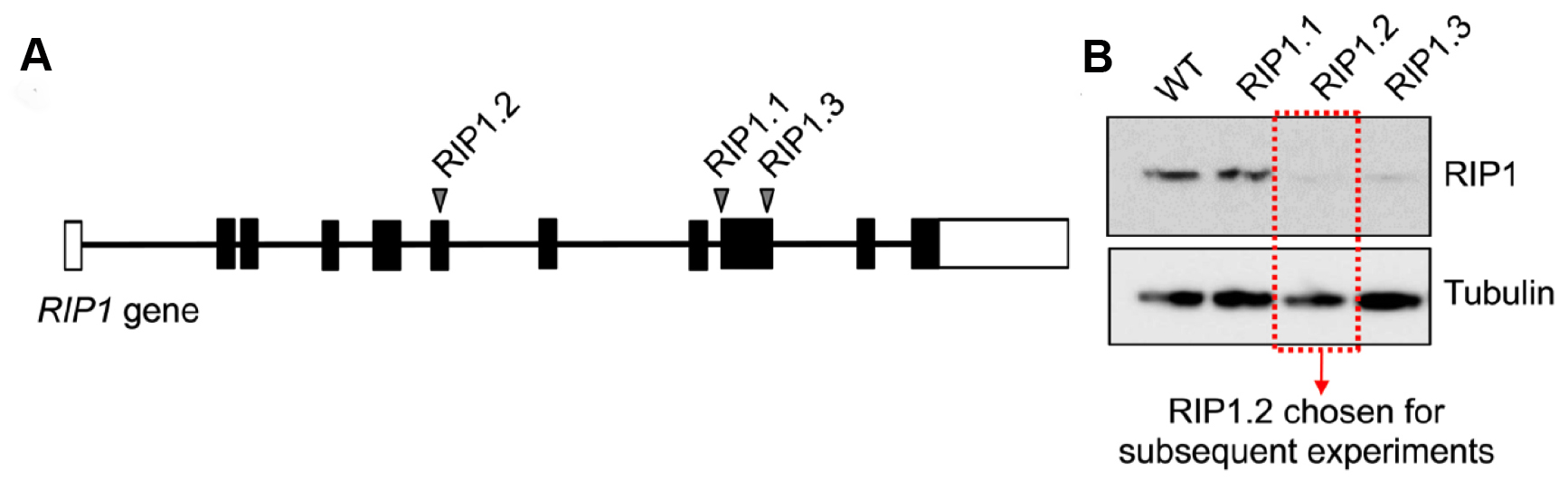
Figure 7. Validation of sgRNA screen for RIP1 knockout in leukemia cell lines. A. Three different sgRNAs were designed for the RIP1 gene. RIP1.2 is located in exon 6, RIP1.1 and RIP1.3 are located in exon 9. Each sgRNA was cloned and transduced via LentiCRISPR viral particles into 1 million cells at an MOI of < 0.1. B. 5 d after viral transduction, cells were sorted for fluorescent signal and cultured for > 2 weeks in vitro to allow gene knockout to occur and generate enough cell material for Western blot. Western blot was developed with mouse anti-RIP1 (1:1,000) and goat anti-mouse-HRP (1:5,000).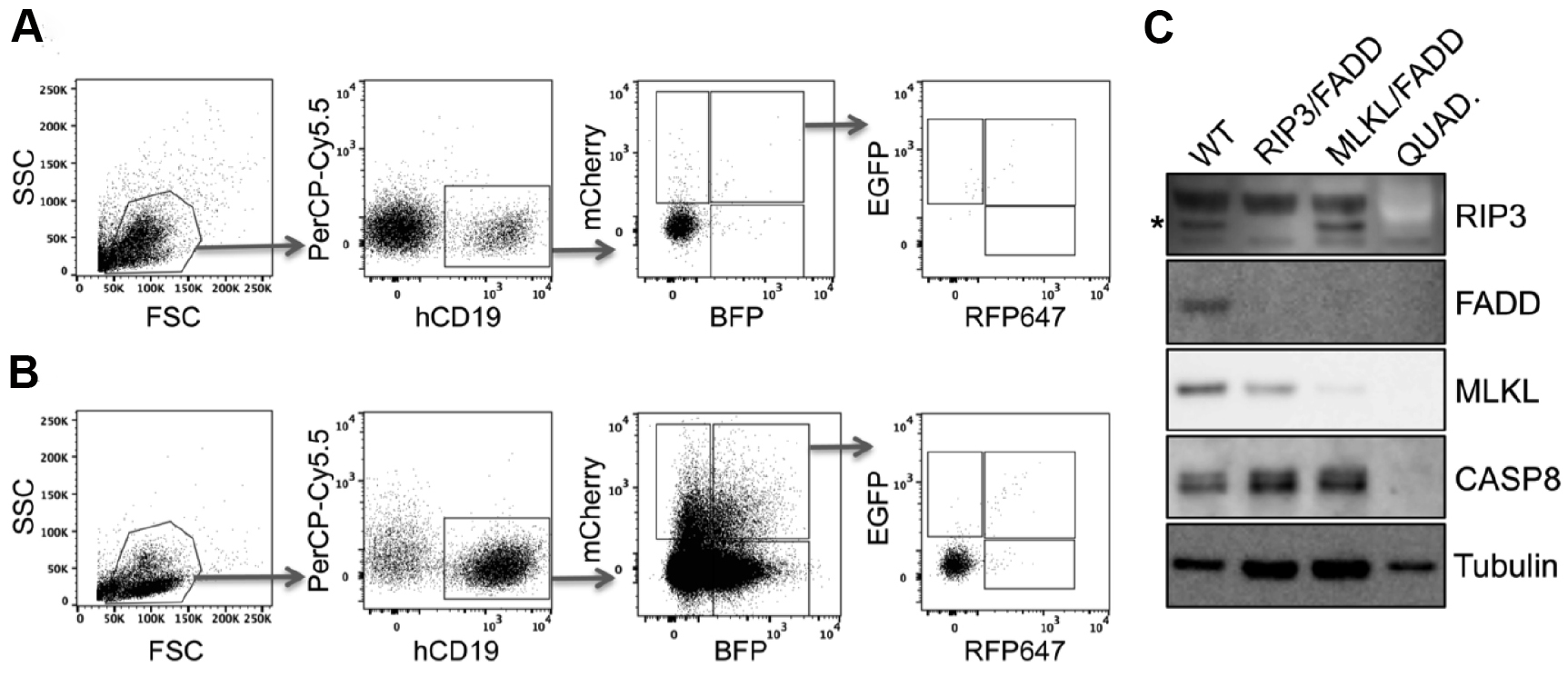
Figure 8. Quadruple knockout in primary human PDX. Primary human acute lymphoblastic leukemia xenografts were transduced either in double or in quadruple combination with RIP3, FADD, MLKL and Caspase8 (CASP8) targeting pLentiCRISPR lentiviral supernatants and directly transplanted into NSG mice. To select for knockout cells mice were treated with birinapant (30 mg/kg) daily (for more details see McComb et al., 2016). A and B. Engraftment was controlled by flow cytometry of peripheral blood. Here we present examples of leukemic engraftment (A) without or (B) with selective birinapant treatment, showing enriched knockout populations. Dot plots show the gating strategy for single and multi-gene knockout cells. First living lymphocytes were defined by forward scatter (FCS) and sideward scatter (SSC). Human engraftment is detected by hCD19 and autofluorescence can be excluded by gating negative cells in an unused channel such as PerCP-Cy5.5 (shown here). Removing autofluorescent cells in this way is extremely helpful for examining rare populations, such as the quadruple positive cells seen here. Subsequently the single or multi-positive cells can be visualized according to their fluorescence (mCherry, BFP, GFP and RFP647). C. To confirm the gene knockout, respective populations were sorted and expanded separately in NSG mice. After harvest, knockout cell lysates were examined via Western blot. Lysates were loaded on two gels in parallel and Western blot was developed in serial detection/stripping steps with following antibodies: rabbit anti-FADD, rat anti-MLKL, rabbit anti-RIP3, mouse anti-CASP8, mouse anti-tubulin, goat anti-mouse-HRP, goat anti-rabbit-HRP, goat anti-rat-HRP. Primary antibody dilution 1:1,000, secondary antibody dilution 1:5,000. *RIP3-specific band.
Notes
Table 2. Troubleshooting advice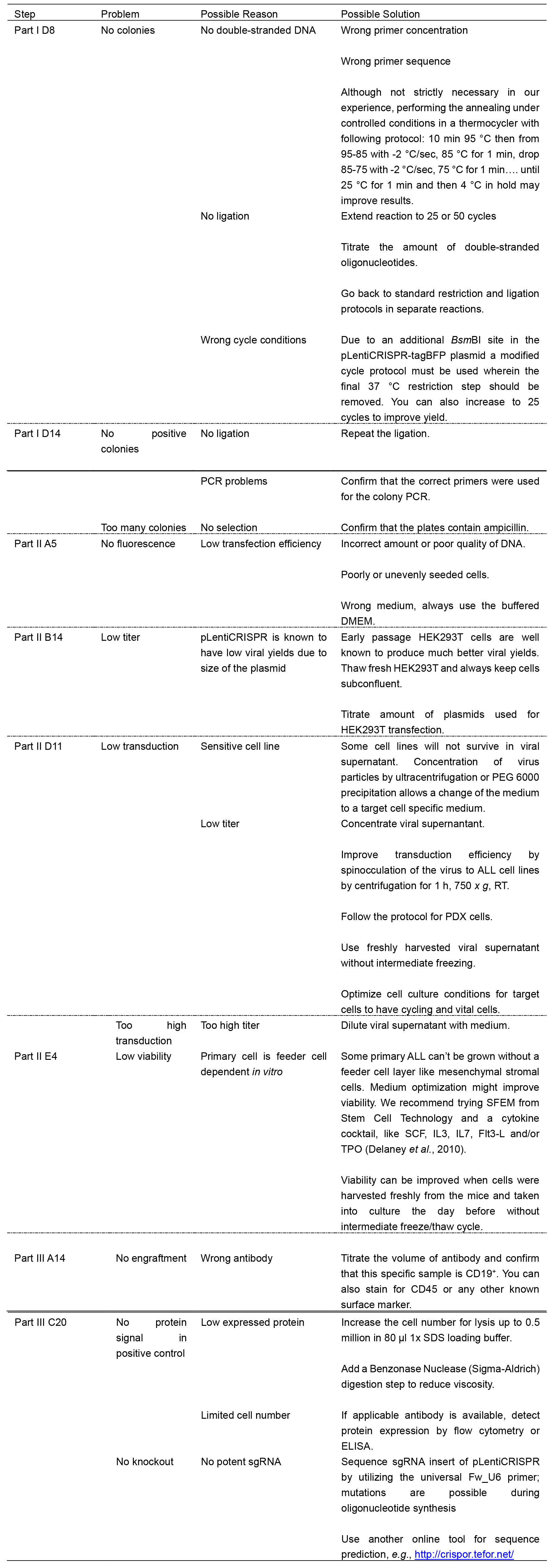
Recipes
- LB agar plates Combine
5 g NaCl
5 g Bacto tryptone
2.5 g yeast extract
7.5 g agar and fill up to 500 ml with ddH2O
Autoclave the solution, cool down, add antibiotics (e.g., Ampicillin 100 µl/ml) and pour into Petri dishes
Wait until the agar solidifies and store at 4 °C - LB medium Combine
5 g NaCl
5 g Bacto tryptone
2.5 g yeast extract and fill up to 500 ml with ddH2O
Autoclave solution, cool down and add antibiotics (e.g., Ampicillin 100 µl/ml)
Store at 4 °C - DMEM complete
Supplement DMEM with 10% (v/v) FCS, 2% (v/v) L-glutamine, 1% (v/v) sodium pyruvate and 2% (v/v) HEPES
Store at 4 °C - RPMI medium
Supplement RPMI with 10% (v/v) FCS, 1% (v/v) L-glutamine and 1% (v/v) Pen/Strep
Store at 4 °C - Freezing medium
Supplement FBS with 10% (v/v) DMSO - 3x SDS lysis buffer
250 mM Trizma HCl pH 6.8
4% (w/v) SDS
0.02% (w/v) bromophenol blue
40% (v/v) glycerol
4% (v/v) 2-mercaptoethanol
To make 1x SDS loading buffer dilute 1:3 in PBS
Aliquots can be stored at -20 °C - Red blood lysis buffer (RBC buffer)
150 mM NH4Cl
8 mM KClO3
0.2 mM EDTA
Mix and filter solution sterile with 0.22 µm filter and aliquot for freezing at -20 °C
CAUTION: Avoid freeze and thaw cycles. Thawed aliquots can be stored at 4 °C for up to one month. - 10x TBS
140 mM NaCl
26 mM KCl
250 mM Trizma base
Adjust pH to 7.4 with HCl
Store at RT - 1x TBS-T
Dilute 10x TBS stock with distilled water and add Tween-20 to 0.1% (v/v)
Store at RT - Ponceau S solution
Dissolve 0.1% (w/v) Ponceau S in 5% (v/v) acetic acid
Store at RT
Solution can be reused - SDS glycine stripping buffer
0.1 M glycine
0.5% (w/v) SDS
Adjust pH to 2.5 (use 12 N HCl)
Store at RT
Acknowledgments
We are indebted to many colleagues for their kind support. Particularly we want to thank B. Marovca for mouse transplantation support, D. Morf and S. Jenni for flow cytometry sorting, C. Stocking (Heinrich-Pette-Institute) for providing her virus production and transduction protocols. This work was supported by the ‘Stiftung Kinderkrebsforschung Schweiz’, the MAM-Fonds of the Children’s Research Centre of the University Children’s Hospital Zurich, the Empiris foundation, the clinical research focus program ‘human hemato-lymphatic diseases’ of the University of Zurich, the Swiss Cancer League (KFS 3609-02-2015), the Novartis Foundation for Biomedical Research, the Swiss national Science Foundation SNF (310030- 133108), the Canadian institutes for health research CIHR, the Forschungskredit of the University of Zurich (FK-14-016) and the Fondation Panacée.
L.H. and S.M. contributed equally to this manuscript. L.H. and S.M. conceived and designed protocols. S.M., J.A. and L.H. performed experiments. Y.H. contributed to the protocol optimization. M.H., A.A. and P.P. developed the single-pot restriction/insertion protocol. L.H., S.M., J.A., JP.B. and B.B. wrote the manuscript with input from all authors. The authors declare that they have no competing financial interests.
The protocol we describe here is based on methods used in our article entitled ‘Activation of concurrent apoptosis and necroptosis by SMAC mimetics for the treatment of refractory and relapsed ALL’ (McComb et al., 2016).
References
- Bétermier, M., Bertrand, P. and Lopez, B. S. (2014). Is non-homologous end-joining really an inherently error-prone process? PLoS Genet 10(1): e1004086.
- Canté-Barrett, K. Mendes, R. D., Smits, W. K., van Helsdingen-van Wijk, Y. M., Pieters, R. and Meijerink, J. P. (2016). Lentiviral gene transfer into human and murine hematopoietic stem cells: size matters. BMC Res Notes 9: 312.
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A. and Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121): 819-823.
- Delaney, C., Heimfeld, S., Brashem-Stein, C., Voorhies, H., Manger, R. L. and Bernstein, I. D. (2010). Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat Med 16(2): 232-236.
- Dever, D. P., Bak, R. O., Reinisch, A., Camarena, J., Washington, G., Nicolas, C. E., Pavel-Dinu, M., Saxena, N., Wilkens, A. B., Mantri, S., Uchida, N., Hendel, A., Narla, A., Majeti, R., Weinberg, K. I. and Porteus, M. H. (2016). 2782 0943" target="_blank">CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature 539(7629): 384-389.
- Gaj, T., Gersbach, C. A. and Barbas, C. F., 3rd (2013). ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31(7): 397-405.
- Hsu, P. D., Scott, D. A., Weinstein, J. A., Ran, F. A., Konermann, S., Agarwala, V., Li, Y., Fine, E. J., Wu, X., Shalem, O., Cradick, T. J., Marraffini, L. A., Bao, G. and Zhang, F. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31(9): 827-832.
- Hsu, P. D., Lander, E. S. and Zhang, F. (2014). Development and applications of CRISPR-Cas9 for genome engineering. Cell 157(6): 1262-1278.
- Ishino, Y., Shinagawa, H., Makino, K., Amemura, M. and Nakata, A. (1987). Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169(12): 5429-5433.
- Jansen, R., Embden, J. D. A. van, Gaastra, W. and Schouls, L. M. (2002). Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43(6): 1565-1575.
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A. and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096): 816-821.
- Kabadi, A. M., Ousterout, D. G., Hilton, I. B. and Gersbach, C. A. (2014). Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res 42(19): e147.
- Kutner, R. H., Zhang, X. Y. and Reiser, J. (2009). Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat Protoc 4(4): 495-505.
- Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., Norville, J. E. and Church, G. M. (2013). RNA-guided human genome engineering via Cas9. Science 339(6121): 823-826.
- Ma, H., Naseri, A., Reyes-Gutierrez, P., Wolfe, S. A., Zhang, S. and Pederson, T. (2015). Multicolor CRISPR labeling of chromosomal loci in human cells. Proc Natl Acad Sci U S A 112(10): 3002-3007.
- McComb, S., Aguade-Gorgorio, J., Harder, L., Marovca, B., Cario, G., Eckert, C., Schrappe, M., Stanulla, M., von Stackelberg, A., Bourquin, J. P. and Bornhauser, B. C. (2016). Activation of concurrent apoptosis and necroptosis by SMAC mimetics for the treatment of refractory and relapsed ALL. Sci Transl Med 8(339): 339ra370.
- Mojica, F. J., Diez-Villasenor, C., Soria, E. and Juez, G. (2000). Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol Microbiol 36(1): 244-246.
- Ousterout, D. G., Kabadi, A. M., Thakore, P. I., Majoros, W. H., Reddy, T. E. and Gersbach, C. A. (2015). Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat Commun 6: 6244.
- Park, C. Y., Sung, J. J., Choi, S. H., Lee, D. R., Park, I. H. and Kim, D. W. (2016). Modeling and correction of structural variations in patient-derived iPSCs using CRISPR/Cas9. Nat Protoc 11(11): 2154-2169.
- Sander, J. D. and Joung, J. K. (2014). CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32(4): 347-355.
- Schmitz, M., Breithaupt, P., Scheidegger, N., Cario, G., Bonapace, L., Meissner, B., Mirkowska, P., Tchinda, J., Niggli, F. K., Stanulla, M., Schrappe, M., Schrauder, A., Bornhauser, B. C. and Bourquin, J. P. (2011). Xenografts of highly resistant leukemia recapitulate the clonal composition of the leukemogenic compartment. Blood 118(7): 1854-1864.
- Shalem, O., Sanjana, N. E., Hartenian, E., Shi, X., Scott, D. A., Mikkelsen, T. S., Heckl, D., Ebert, B. L., Root, D. E., Doench, J. G. and Zhang, F. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343(6166): 84-87.
- Shi, J., Wang, E., Milazzo, J. P., Wang, Z., Kinney, J. B. and Vakoc, C. R. (2015). Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol 33(6): 661-667.
- Tiscornia, G., Singer, O. and Verma, I. M. (2006). Production and purification of lentiviral vectors. Nat Protoc 1(1): 241-245.
- Wang, F. and Qi, L. S. (2016). Applications of CRISPR genome engineering in cell biology. Trends Cell Biol 26(11): 875-888.
- Weber, K., Thomaschewski, M., Benten, D. and Fehse, B. (2012). RGB marking with lentiviral vectors for multicolor clonal cell tracking. Nat Protoc 7(5): 839-849.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Behrmann, L., McComb, S., Aguadé-Gorgorió, J., Huang, Y., Hermann, M., Pelczar, P., Aguzzi, A., Bourquin, J. P. and Bornhauser, B. C. (2017). Efficient Generation of Multi-gene Knockout Cell Lines and Patient-derived Xenografts Using Multi-colored Lenti-CRISPR-Cas9. Bio-protocol 7(7): e2222. DOI: 10.21769/BioProtoc.2222.
Category
Molecular Biology > DNA > Mutagenesis
Cell Biology > Cell isolation and culture > Cell isolation > Flow cytometry
Cell Biology > Cell engineering > CRISPR-cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.