- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Next-generation Sequencing of the DNA Virome from Fecal Samples


Published: Vol 7, Iss 5, Mar 5, 2017 DOI: 10.21769/BioProtoc.2159 Views: 11260
Reviewed by: Yannick DebingAnca Flavia SavulescuYi Zhang
Original research article
The authors used this protocol in:

Mar 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Herein we describe a detailed protocol for DNA virome analysis of low input human stool samples (Monaco et al., 2016). This protocol is divided into four main steps: 1) stool samples are pulverized to evenly distribute microbial matter; 2) stool is enriched for virus-like particles and DNA is extracted by phenol-chloroform; 3) purified DNA is multiple-strand displacement amplified (MDA) and fragmented; and 4) libraries are constructed and sequenced using Illumina Miseq. Subsequent sequence analysis for viral sequence identification should be sensitive but stringent.
Keywords: ViromeBackground
The virome, a dynamic community of eukaryotic viruses, bacteriophages and endogenous retroviruses, represents a minimally characterized component of the human microbiome (Virgin, 2014). In fact, it is estimated that only 1% of the virome has been sequenced and annotated (Mokili et al., 2012). Next generation sequencing (NGS) enables examination of the entire virome, including unculturable viruses. Stool is a readily obtainable specimen type for study of the virome, and alterations in the fecal virome have been associated with a number of disease states (Handley et al., 2012; Norman et al., 2015; Monaco et al., 2016). The fecal virome is largely comprised of bacteriophages, which affect the gastrointestinal tract through alterations in bacterial functions and populations (Duerkop and Hooper, 2013; Reyes et al., 2013; Virgin, 2014). Enteric eukaryotic viruses, while less ubiquitous than bacteriophages, play a more direct role in gastrointestinal tract dysfunction by inducing gastroenteritis, enteritis and colitis. Despite the abundance of bacteriophages in fecal samples, only a few studies thus far have examined the contributions of fecal bacteriophages in human diseases. Inflammatory bowel disease has been associated with increased enteric bacteriophage richness (Norman et al., 2015). In contrast, profound immunosuppression from AIDS in a sub-Saharan cohort resulted in an expanded eukaryotic virome, but had minimal impact on bacteriophage populations (Monaco et al., 2016). More studies are needed to elucidate the role the fecal virome plays in disease states. A key roadblock to studying the stool virome is viral nucleic acid extraction and enrichment from fecal material. Several factors can contribute to difficulty in isolating viral sequences from fecal samples, chief among them the fact that viruses constitute a minority of fecal sample material. Additionally, dilution of feces in collection media (such as RNAlater RNA stabilization reagent) can further hamper the ability to find viral sequences. While many nucleic acid extraction protocols can be used for high input nucleic acid samples to enrich for viral nucleic acid, low input samples, such as those diluted in collection media, represent a challenge with virome studies. After comparison and optimization of several methods, the following protocol was identified as the most universally applicable for isolation of phage and DNA viral sequences from both low (Monaco et al., 2016) and high (Norman et al., 2015) input samples.
Materials and Reagents
- Stool aliquoting and pulverization
- Versi-dry sheets (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 74018-00 )
- Large Kim-wipes (KCWW, Kimberly-Clark, catalog number: 34721 )
- Extra heavy-duty aluminum foil (VWR, catalog number: 89107-734 )
- Dry ice
- Liquid nitrogen and a Dewar along with Styrofoam cup
- Pre-labeled screw capped tubes (STARSTEDT) in a freezer box (4 pre-labeled tubes per sample)
- Stool scrapers (autoclaved in sets of 6) (Fisher Scientific, catalog number: 21-401-25B )
- Sterilization pouches (small)
- Autoclave bags
- Bleach (5.25% solution of sodium hypochlorite)
- 75% EtOH
- Versi-dry sheets (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 74018-00 )
- Virus-like particle (vlp) preparation
- Sterile 1.5 ml and 2.0 ml screw-cap tubes
- 1 ml luer-lok syringes (BD, catalog number: 309628 )
- 0.45 µm filters 13 mm diameter (EMD Millipore, catalog number: SLHV013SL )
- 0.22 µm filters 13 mm diameter (EMD Millipore, catalog number: SLGV013SL )
- 1 M Tris, pH 7.5 (Fisher Scientific, catalog number: MT-46-030-CM ) (Tris 1 M pH 7.5, DNase-, RNase-, protease-free [6 x 1 L bottles])
- 5 M NaCl (Promega, catalog number: V4221 ) (5 M NaCl 1 L bottle, DNase-, RNase-, protease-free; aliquoted in 1 ml aliquots and stored at -20 °C)
- Saline magnesium (SM) buffer (Fisher Scientific, catalog number: 50-329-444 ) (SM buffer with gelatin pH 7.5, 100 ml)
- 10% SDS (diluted from 20% SDS stock [Thermo Fisher Scientific, AmbionTM, catalog number: AM9820 ] in RNase, DNase, Protease free H2O; aliquoted and stored at -20 °C)
- Lysozyme (10 mg/ml) (EMD Millipore, catalog number: 71412 ) – aliquoted and stored at -20 °C
- Turbo DNase I (2 U/µl) (Thermo Fisher Scientific, AmbionTM, catalog number: AM2238 )
- BaseLine zero DNase (1 U/µl) (Epicentre, catalog number: DB0711K )
- Chloroform (Fisher Scientific, catalog number: C298-500 )
- Phenol:chloroform:isoamyl alcohol (25:24:1) pH 8.0 (Fisher Scientific, catalog number: BP1752I-100 )
- QIAGEN DNeasy Blood and Tissue Kit (QIAGEN, catalog number: 69506 )
- CTAB (Sigma-Aldrich, catalog number: 52365 )
- CTAB/NaCl (see Recipes) [0.45 µm filtered + 0.22 µm filtered]
- Sterile 1.5 ml and 2.0 ml screw-cap tubes
- Library construction
- GenomiPhi V2 DNA Amplification Kit (GE Healthcare, catalog number: 25-6600-31 )
- Covaris microTUBE AFA fiber snap-cap 50 µl (Covaris, catalog number: 520045 )
- NEBNext® UltraTM DNA Library Prep Kit for Illumina (New England Biolabs, catalog number: E7370L , 96 rxns)
- NEBNext® Multiplex Oligos for Illumina®
Index Primers Set 1 (New England Biolabs, catalog number: E7335L )
Index Primers Set 2 (New England Biolabs, catalog number: E7500L ) - AMPure XP beads (Beckman Coulter, catalog number: A63881 )
- TE low EDTA
- D1K reagents (Agilent Technologies, catalog number: 5067-5362 )
- D1K screen tape for TapeStation (Agilent Technologies, catalog number: 5067-5361 )
- GenomiPhi V2 DNA Amplification Kit (GE Healthcare, catalog number: 25-6600-31 )
Equipment
- Biosafety hood
- 6 mortar/pestles, 100 ml capacity
- Beaker
- Microcentrifuge
- PCR hood
- -80 °C freezer
- Covaris E210
- PCR Thermocycler (Eppendorf)
- NanoDrop Micro-Volume UV-Vis spectrophotometer
- Agilent 2200 TapeStation (Agilent Technologies, model: 2200 TapeStation)
- Loading tips for TapeStation (Agilent Technologies, catalog number: 5067-5153 )
- DynamagTM-spin magnet (Thermo Fisher Scientific, catalog number: 12320D )
Procedure
- Stool aliquoting/pulverization
Note: All steps were performed in a biosafety hood due to use of human feces and risk of viable infectious organisms.- Washing and preparation of mortars and pestles
- Thoroughly sterilize working area with 10% bleach followed by 75% EtOH and UV exposure for 10 min to minimize contamination.
- Prepare 10% bleach beaker for scrapers.
- Bleach mortar/pestles for 10 min in 10% bleach solution.
- Place versi-dry sheets in hood.
- Place samples on dry ice in hood.
- With remaining dry ice, place open microtube freezer box stably on top of dry ice with empty pre-labeled tubes and place in hood.
- Tear heavy duty aluminum foil into pieces around 10 inches wide. Then using a flat surface, tear this into 3 approximately equal squares. Do this twice (6 squares). Place these in the hood.
- Once the 10 min bleach is finished, rinse all 6 mortars and pestles with dH2O and set on another versi-dry to dry slightly, and then set upright in hood on versi-dry. UV for 10 min along with all other equipment to be used.
- When done, place pestles in mortars, and set all but one to side, stack aluminum foil.
- Add large mound of gloves to hood.
- Add liquid N2 to Dewar.
- Pulverization of stool
- Double glove.
Note: This is not needed if not using human stool. - Place large Kim-wipe on top of the versi-dry. Place mortar and pestle on it.
- Open a sterile package of stool scrapers.
- Place a piece of aluminum foil into mortar and mold to inner shape of mortar.
- Select sample to use, bring out of dry ice and set on Kim-wipe.
- Identify corresponding pre-labelled screw-cap tubes, and bring them to front of the box.
- Fill Styrofoam cup 3/4 full with liquid N2 from Dewar. Pour some liquid N2 into the box of screw-top tubes, and pour enough into the mortar to fill it ½ way full. Place cup to side of mortar (recommend having some liquid N2 remaining).
- Take a stool scraper out of package.
- Open sample tube.
- Use stool scraper to carefully get sample out of tube into the mortar. Try to get the whole sample out at once.
Note: May need to warm sample tube in hands to slightly thaw around edges. Do not thaw longer than necessary to remove sample. - Once out, place now empty sample tube into the box of tubes to re-freeze, consider adding small amount of liquid N2 near the tube to aid with re-freezing.
- For large chunks of stool, use pestle to break apart. When all large chunks have been crushed to small pieces, then pulverize with pestle. Intermittently add more liquid N2 to mortar as needed to keep sample from thawing out until pulverized completely.
- Let liquid N2 boil off. Sample now has the texture of a powder.
- Immediately take a fresh tube and scrape approximately 200 mg sample into the tube, tap the tube onto clean side of cold mortar to settle contents to bottom, and place it back into microtube box. Repeat with the remaining 3 tubes. If any sample remains after aliquoting 200 mg to new tubes, scrape remaining sample into the original tube, tapping tube onto a clean cold portion of the mortar to settle contents to the bottom of the tube. If needed, remove aluminum foil from the mortar to get any remaining bits of sample.
Notes:
a. Sample left in aluminum foil thaws VERY quickly once out of the mortar.
b. Do not screw on caps tightly as it is likely they still have some liquid N2 present. - Place stool scraper into bleach beaker, aluminum foil into autoclave bag, pestle into mortar, deglove, take Styrofoam cup out of hood. Place Kim-wipe into an autoclave bag. If versi-dry is dirty, remove versi-dry and place it into the autoclave bag.
- Carefully place mortar and pestle in bleach bath.
- Repeat process for total of 6 samples.
- Store samples at -80 °C.
- Wash as above and repeat as needed.
- Washing and preparation of mortars and pestles
- VLP enrichment protocol
- Pulverize as above ~200 mg of stool into sterile 2 ml screw cap tube on dry ice to keep frozen.
- Add 400 µl cold SM buffer per sample. Keep on ice. Vortex on high speed for 5 min.
Note: It is possible to use a 2 ml tube adapter plate to hold all sample tubes at once if available. Avoid touching tip to tube when adding reagents. Change tips between samples to avoid cross-contamination. Low speed centrifugation is used to avoid lysing bacteria. Goal is to get everything into solution. More SM buffer can be added if needed. - Centrifuge for 10 min at 2,000 x g at 4 °C. Transfer supernatant to a clean 1.5 ml Eppendorf tube. Centrifuge a second time for 10 min at 2,000 x g at 4 °C.
Note: Sometimes a third spin is needed to clarify supernatant. Final supernatant volume should be at least 200 µl because a fraction of the volume will be lost in the subsequent filtration. More SM buffer can be added if needed. - Filter supernatant once through a 0.45 µm filter. Then filter twice through 0.22 µm filters. All filtration steps use 1 ml Luer-lok syringes and 13 mm diameter filters.
Note: Filtering once through 0.22 µm filter is sufficient, but 2 x 0.22 µm filtration steps were used in this manuscript. If the filter clogs: pull the plunger out to relieve pressure, take off the filter and put a new one on. If the initial sample volume was low, may need to use a pipette to recover remaining sample from the old, clogged filter. - Check final volume of filtrate. Bring to 200 µl with cold SM buffer.
- Take 200 µl to a clean 1.5 ml tube on ice.
- Store any remainder for later use at -80 °C.
- Lysozyme/chloroform/DNase treatment
- Add 20 µl (10% volume) lysozyme (10 mg/ml stock) to each tube. Incubate at 37 °C 30 min.
- Add 44 µl (20% volume) chloroform, briefly vortex, and incubate 10 min at room temperature (RT; 15-25 °C).
- Centrifuge at 2,500 x g 5 min at RT.
- Collect aqueous phase and transfer to 2 ml screw cap tube.
Note: Screw cap is preferable to prevent sample loss from the top popping open during the heating. - Make DNase master mix.
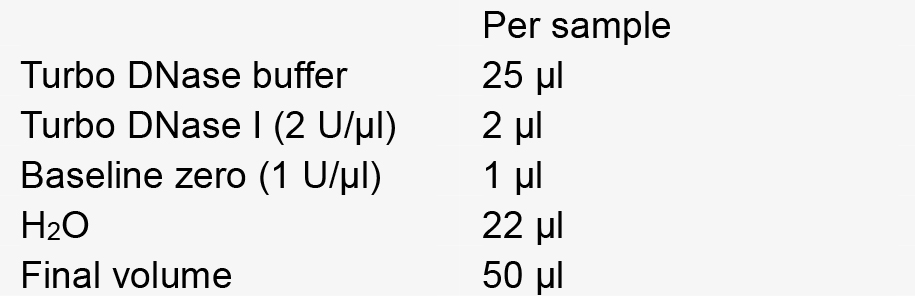
- Add 20 µl (10% volume) lysozyme (10 mg/ml stock) to each tube. Incubate at 37 °C 30 min.
- Add 50 µl DNase master mix to each tube. Incubate at 37 °C for 1 h.
- Heat-inactivate DNase at 65 °C for 15 min followed by a quick spin to pull down condensation.
- SDS/CTAB cleanup
- Add 10 µl SDS (10%) + 1 µl proteinase K (10 mg/ml stock) to each tube and incubate at 56 °C for 20 min. At this point, pre-incubate CTAB/NaCl solution at 65 °C.
- Add 35 µl NaCl (5 M soln) + 28.1 µl CTAB/NaCl (2.5% soln). Pulse vortex. Incubate at 65 °C for 10 min then perform a quick spin.
Note: Sample will turn cloudy after CTAB/NaCl is added. - Add 200 µl phenol:chloroform:isoamyl alcohol (25:24:1) pH 8.0. Pulse vortex. Centrifuge 8,000 x g for 5 min at RT.
- Collect the aqueous fraction from step B9c. Add 200 µl chloroform. Pulse vortex for 3-5 sec. Centrifuge 8,000 x g for 5 min at RT.
- Collect the aqueous fraction from step B9d. This is final Virus Nucleic Acid.
- Place in dry ice for transport and store at -80 °C.
- Clean DNA using QIAGEN DNeasy Blood and Tissue Kit. Elute in 200 μl elution buffer.
Note: Start at the buffer AL step in the Purification of Total DNA from Animal Blood or Cells protocol. Other column clean-up kits may be used, but should remain consistent between samples used for comparison. We would recommend only clean-up kits that have a large size range of DNA retained. - Add 200 μl buffer AL. Mix thoroughly by vortexing.
- Add 200 μl ethanol (96-100%). Mix thoroughly by vortexing.
- Pipet 650 µl of mixture into a DNeasy mini spin column placed in a 2 ml collection tube. Centrifuge at ≥ 6,000 x g for 1 min. Discard the flow-through. Repeat as necessary until all of sample is used.
- Add 500 μl buffer AW1. Centrifuge for 1 min at ≥ 6,000 x g. Discard the flow-through.
- Add 500 μl buffer AW2, and centrifuge for 2 min at 20,000 x g. Discard the flow-through and collection tube.
- Centrifuge for 1 min at 20,000 x g in a new collection tube.
- Transfer the DNeasy spin column to a new 1.5 ml or 2 ml microcentrifuge tube.
- Elute the DNA by adding 200 μl buffer AE to the center of the DNeasy spin column membrane. Incubate for 1 min at RT. Centrifuge for 1 min at ≥ 6,000 x g.
- Recirculate the eluate through the column once (Add eluate to the center of the DNeasy spin column membrane. Incubate for 1 min at RT. Centrifuge for 1 min at ≥ 6,000 x g).
- Pulverize as above ~200 mg of stool into sterile 2 ml screw cap tube on dry ice to keep frozen.
- Phi29 polymerase DNA amplification
Note: 2 μl of each sample is used as template in 4 independent MDA reactions using the GenomiPhi V2 DNA Amplification Kit to reduce amplification bias. The 4 replicates are then pooled after MDA and quantified by NanoDrop.- Add 8 µl of GenomiPhi V2 DNA Amplification Kit sample buffer + 2 μl of template to a clean 0.5 ml PCR tube.
- Heat at 95 °C for 3 min then cool to 4 °C on ice. Keep cold.
- Prepare master mix on ice:

- Add 10 µl of the above master mix per tube and return to thermocycler.
- Heat to 30 °C for 2 h. Then heat-kill enzyme at 65 °C for 10 min. Cool to 4 °C. Can store at -20 °C.
Note: Incubation time should be optimized to the shortest time that allows sufficient amplification to minimize amplification bias. - Pool the 4 independent MDA reactions from the same sample. Adjust volume to 200 µl with DNase-free H2O.
- Purify MDA product using QIAGEN DNeasy Blood and Tissue Kit. Elute in 100 µl buffer AE, re-circulate through column once to increase yield.
- Add 200 μl buffer AL. Mix thoroughly by vortexing.
- Add 200 μl ethanol (96-100%). Mix thoroughly by vortexing.
- Pipet the mixture into a DNeasy mini spin column placed in a 2 ml collection tube. Centrifuge at ≥ 6,000 x g for 1 min. Discard the flow-through.
- Add 500 μl buffer AW1. Centrifuge for 1 min at ≥ 6,000 x g. Discard the flow-through.
- Add 500 μl buffer AW2, and centrifuge for 2 min at 20,000 x g. Discard the flow-through and collection tube.
- Centrifuge for 1 min at 20,000 x g.
- Transfer the spin column to a new 1.5 ml or 2 ml microcentrifuge tube.
- Elute the DNA by adding 100 μl buffer AE to the center of the spin column membrane. Incubate for 1 min at RT. Centrifuge for 1 min at ≥ 6,000 x g.
- Recirculate the eluate through the column once.
- Check the concentration by NanoDrop.
- Add 8 µl of GenomiPhi V2 DNA Amplification Kit sample buffer + 2 μl of template to a clean 0.5 ml PCR tube.
- Covaris fragmentation with Covaris E210
- Dilute 200 ng DNA to 50 µl total volume dH2O per sample in the Covaris snap-top tubes.
Note: Increasing to 500 ng DNA at this step did not increase number of different viral sequences. - Pre-chill the Covaris.
- Fragment nucleic acid using the 400 setting as per Covaris manual: Intensity 5, Duty cycle 5%, Cycles per burst 200, Treatment time 55 sec, Temp 7 °C, Water level 6, Sample volume 50 µl.
- Remove samples and store on ice if proceeding immediately to the next step (recommended) or store at -20 °C for later use.
- Dilute 200 ng DNA to 50 µl total volume dH2O per sample in the Covaris snap-top tubes.
- NEBNext DNA library construction
Note: Perform as per NEBNext® UltraTM DNA Library Prep Kit for Illumina manual protocol.- End repair
- Bring DNA up to 55.5 µl with dH2O.
- Add 6.5 µl End repair reaction buffer.
- Add 3 µl End Prep enzyme mix.
- In thermocycler:
20 °C for 30 min
65 °C for 30 min
4 °C Hold
- Bring DNA up to 55.5 µl with dH2O.
- Adaptor ligation
- Add 15 µl Blunt/TA ligase master mix.
- Add 2.5 µl undiluted NEBNext adaptor.
- Add 1 µl ligation enhancer.
- In thermocycler, incubate at 20 °C for 15 min.
- Add 3 µl USER enzyme to ligation mix and heat in thermocycler at 37 °C for 15 min.
- AmPure bead size selection for 400-500 bp library product (300-400 bp insert).
- Add 13.5 µl nuclease-free H2O to the ligation reaction for a final volume of 100 µl.
- Add 40 µl AmPure beads and mix well by pipetting up and down.
- Incubate at RT for 5 min.
- Centrifuge tubes briefly. Place on magnet and incubate at RT for 5 min (until clear).
- Transfer supernatant to a clean tube.
- Add 20 µl AmPure beads and mix well. Incubate at RT for 5 min.
- Centrifuge tubes briefly. Place on magnet and incubate at RT for 5 min.
- Leaving tubes on magnet, remove and discard supernatant, careful to not dislodge beads.
- With tubes on magnet, add 200 µl freshly prepared 80% EtOH and incubate at RT for 30 sec.
- With tubes on magnet, remove EtOH wash.
- Repeat steps E4i and E4j two more times for a total of 3 washes.
- Air dry beads for 10 min at RT on the magnet with the tube top open.
- Remove tubes from magnet and add 28 µl TE low EDTA, pH 8 to beads and resuspend by vortexing or mixing well by pipetting.
- Centrifuge tubes briefly, replace tubes on magnet and incubate at RT for 5 min, or until clear.
- Collect 23 µl of DNA and transfer to a clean PCR tube. Be sure not to transfer any beads.
- Add 13.5 µl nuclease-free H2O to the ligation reaction for a final volume of 100 µl.
- PCR amplification
- Add the following to each tube:

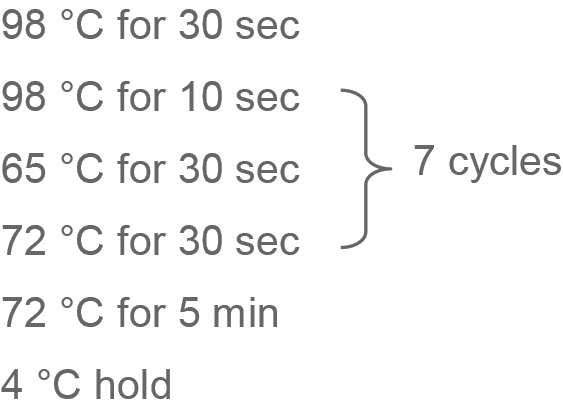
- In a thermocycler:
- PCR cleanup
- Add 50 µl AmPure beads and mix well.
- Incubate at RT x 5 min.
- Centrifuge tubes briefly, place on magnet and incubate at RT for 3-5 min.
- Leaving tubes on magnet, remove supernatant, careful to not dislodge beads.
- With tubes on magnet, add 200 µl freshly prepared 80% EtOH and incubate at RT for 30 sec.
- With tubes on magnet, remove EtOH wash.
- Repeat steps E6e and E6f once.
- Air dry beads for 10 min at RT.
- Remove tubes from magnet and add 33 µl TE low EDTA to beads and resuspend. Incubate at RT for 2 min.
- Replace tubes on magnet and incubate at RT for 4 min.
- Collect 30 µl of DNA and transfer to a clean PCR tube.
- Assay for library yield and quality on TapeStation HS DNA as per TapeStation HS DNA protocol.
- Pool samples (~12 samples/run, equimolar) to a final concentration of 10 nM and verify concentration on TapeStation HS DNA.
- Submit for Illumina Miseq as per facility protocol and recommendations (we used loading concentration 7 pM, 1% PhiX spike-in, Std flowcell, 2 x 250 bp run).
Note: This protocol is also applicable for Illumina Hiseq.
- End repair
Data analysis
Sequence analysis methods are rapidly evolving due to advances in both hardware speed and software coding. Many sequence processing software tools are open-source (such as BBTools, http://jgi.doe.gov/data-and-tools/bbtools/), as are statistical analysis and graphing packages in R (https://www.r-project.org/). We used VirusSeeker (Zhao et al., 2017), a customized automated bioinformatics pipeline based on VirusHunter (Zhao et al., 2013), to detect sequences sharing nucleotide and amino acid sequence similarity to known viruses (Figure 1 below). We recommend using a stringent protocol for viral sequence identification, such as VirusSeeker, that removes low-quality sequences, repeat sequences, and non-specific viral ‘hits.’ Similarly stringent methods have identified novel viral sequences (Zhao et al., 2013). Basic steps in the analysis protocol are shown in Figure 1. Custom viral databases can be generated after downloading sequences corresponding to all viral genomes from the NCBI database (make note of the date of download as new sequences are frequently added). Deduplication is recommended to minimize amplification bias, and taxon-assigned sequences should be normalized to account for variations in sequencing depth between samples. Novel viral sequences identified or viral sequences of interest should be validated by real-time qPCR using primers specific to the viral sequence. Additionally, sequences can be de novo assembled into longer contigs and compared to the NCBI nr/nt databases to better identify phylogeny of viral sequences of interest (Monaco et al., 2016). Phylogenetic trees comparing sequences of interest to known related viral sequences can be made using free software such as FigTree (http://tree.bio.ed.ac.uk/software/figtree/).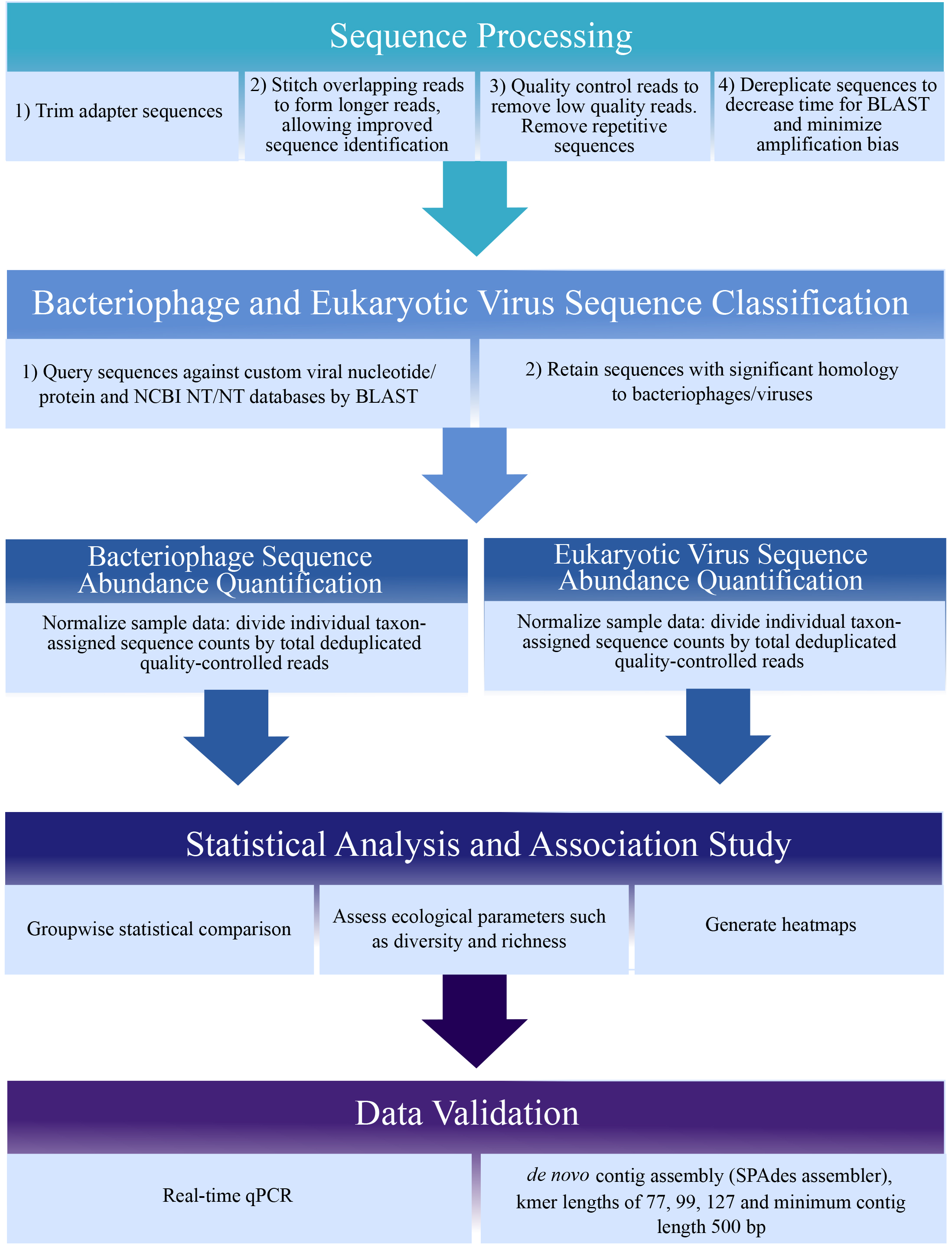
Figure 1. Sequence analysis schematic
Notes
Stool pulverization is performed in order to evenly distribute microbial matter in the sample. The use of SM buffer containing gelatin stabilizes bacteriophage populations after freezing for further characterization of bacteriophage of interest, including culturing. Due to risk of contamination, reagents should be used only for fecal microbiome studies. Use the same reagents for all Illumina Miseq runs (i.e., same bottle of sterile water, kits, etc.) as many reagents may be contaminated by microbial DNA, and this ensures even contamination across runs.
Recipes
- CTAB/NaCl
3.5 ml 5 M NaCl (final conc. 0.7 M)
2.5 g CTAB (final conc. 10%)
12 ml nuclease-free H2O
Final volume: 25ml
Heat to 55 °C to dissolve
Add water to final volume of 25 ml. Then 0.45 µm followed by 0.22 µm filter
Acknowledgments
CLM was supported by NIH training grant 5T32AI007172-34, and the work was funded by R24 ODO19793, R01 OD011170, R01 AI111918, and R01 DK101354. DSK is supported by the Burroughs Wellcome Fund. The stool pulverization protocol is adapted from a protocol generously provided by the Jeffrey Gordon laboratory at Washington University in St. Louis. The VLP protocol was adapted from a protocol for phage isolation (Reyes et al., 2013). We would like to thank Brian Keller, M.D., Ph. D for critical review of this manuscript.
References
- Duerkop, B. A. and Hooper, L. V. (2013). Resident viruses and their interactions with the immune system. Nat Immunol 14(7): 654-659.
- Handley, S. A., Thackray, L. B., Zhao, G., Presti, R., Miller, A. D., Droit, L., Abbink, P., Maxfield, L. F., Kambal, A., Duan, E., Stanley, K., Kramer, J., Macri, S. C., Permar, S. R., Schmitz, J. E., Mansfield, K., Brenchley, J. M., Veazey, R. S., Stappenbeck, T. S., Wang, D., Barouch, D. H. and Virgin, H. W. (2012). Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome. Cell 151(2): 253-266.
- Mokili, J. L., Rohwer, F. and Dutilh, B. E. (2012). Metagenomics and future perspectives in virus discovery. Curr Opin Virol 2(1): 63-77.
- Monaco, C. L., Gootenberg, D. B., Zhao, G., Handley, S. A., Ghebremichael, M. S., Lim, E. S., Lankowski, A., Baldridge, M. T., Wilen, C. B., Flagg, M., Norman, J. M., Keller, B. C., Luevano, J. M., Wang, D., Boum, Y., Martin, J. N., Hunt, P. W., Bangsberg, D. R., Siedner, M. J., Kwon, D. S. and Virgin, H. W. (2016). Altered virome and bacterial microbiome in human immunodeficiency virus-associated acquired immunodeficiency syndrome. Cell Host Microbe 19(3): 311-322.
- Norman, J. M., Handley, S. A., Baldridge, M. T., Droit, L., Liu, C. Y., Keller, B. C., Kambal, A., Monaco, C. L., Zhao, G., Fleshner, P., Stappenbeck, T. S., McGovern, D. P., Keshavarzian, A., Mutlu, E. A., Sauk, J., Gevers, D., Xavier, R. J., Wang, D., Parkes, M. and Virgin, H. W. (2015). Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 160(3): 447-460.
- Reyes, A., Wu, M., McNulty, N. P., Rohwer, F. L. and Gordon, J. I. (2013). Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proc Natl Acad Sci U S A 110(50): 20236-20241.
- Virgin, H. W. (2014). The virome in mammalian physiology and disease. Cell 157(1): 142-150.
- Zhao, G., Krishnamurthy, S., Cai, Z., Popov, V. L., Travassos da Rosa, A. P., Guzman, H., Cao, S., Virgin, H. W., Tesh, R. B. and Wang, D. (2013). Identification of novel viruses using VirusHunter--an automated data analysis pipeline. PLoS One 8(10): e78470.
- Zhao, G., Wu, G., Lim, E.S., Droit, L., Krishnamurthy, S., Barouch, D.H., Virgin, H.W., and Wang, D. (2017). VirusSeeker, a computational pipeline for virus discovery and virome composition analysis. Virology 503: 21-30.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Monaco, C. L. and Kwon, D. S. (2017). Next-generation Sequencing of the DNA Virome from Fecal Samples. Bio-protocol 7(5): e2159. DOI: 10.21769/BioProtoc.2159.
Category
Microbiology > Microbial genetics > DNA > DNA sequencing
Molecular Biology > DNA > Genotyping
Systems Biology > Genomics > Sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.