- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Chromatographic Separation of the Codonocarpine Type Alkaloids from the Root Bark of Capparis decidua



Published: Vol 7, Iss 4, Feb 20, 2017 DOI: 10.21769/BioProtoc.2144 Views: 8484
Reviewed by: Arsalan DaudiMing LuoMagdalena Migocka
Original research article
The authors used this protocol in:

Aug 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Various parts of the caper tree Capparis decidua have found application in traditional medicine. The isolation and structural elucidation of the codonocarpine type alkaloids contained in the root bark, however, is not trivial and has probably led to misinterpretation in the past. This protocol describes the extraction and chromatographic separation of the four major alkaloids of the root bark of Capparis decidua. The delivered samples of cadabicine, codonocarpine, isocodonocarpine and capparidisinine were suitable for their unambiguous structural elucidation by NMR, MS and MS/MS.
Keywords: Capparis deciduaBackground
The tree Capparis decidua is widely distributed in the arid regions of Africa, the Middle East and Southern Asia, where various parts of the plant are commonly used in local folk medicines for the treatment of various disorders. The root bark, for example, is applied as anthelmintic and purgative, and it has been shown that its alcoholic extract possesses significant antibacterial and antifungal activities (Singh et al., 2011; Singh and Singh, 2011; Tlili et al., 2011; Mohammed et al., 2015).
Ahmad et al. (1985; 1986; 1987; 1989; Arif, 1986) intensively studied the root bark extract of Capparis decidua and published several structures of codonocarpine type alkaloids. Some of these structures did not fit with our understanding of the biosynthesis of such alkaloids. Further, we were not convinced that the given analytic data and its interpretation gives unambiguous proof for the structural claims (Bienz et al., 2002). Therefore, we initiated our own investigation. We developed a protocol for the extraction and chromatographic separation of codonocarpine type alkaloids and could isolate three alkaloid fractions that allowed the identification and structural elucidation of the four major spermidine alkaloids of the root bark of Capparis decidua: cadabicine (1), codonocarpine (2), isocodonocarpine (3), and capparidisinine (4) (Figure 1) (Forster et al., 2016). 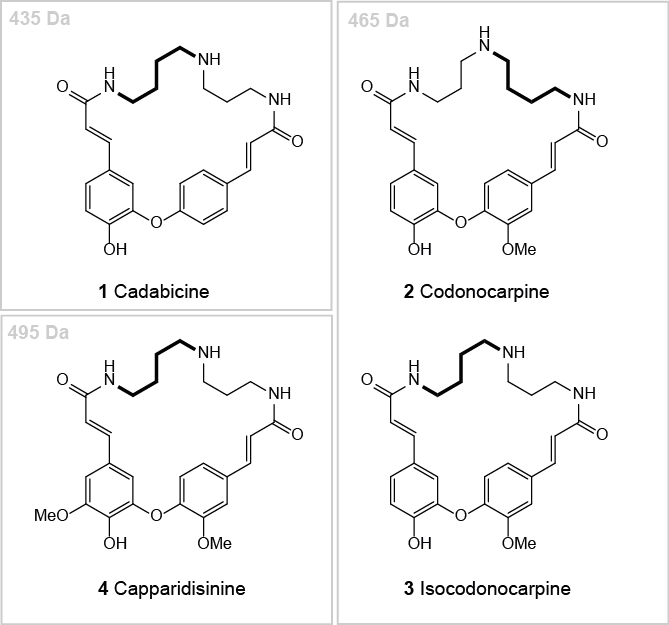
Figure 1. The four major codonocarpine type alkaloids found in the root bark of Capparis decidua. (1) Cadabicine; (2) Codonocarpine; (3) Isocodonocarpine; and (4) Capparidisinine.
The isolation and structure elucidation of alkaloids is not trivial. The class of alkaloids, defined as nitrogen containing organic compounds from natural sources with a most basic character, comprises more than 10,000 compounds. As the properties and structures of alkaloids differ strongly from compound to compound, there is no general way to isolate them. Nevertheless, most alkaloids exist naturally in their protonated form. Hence, the methanolic extraction of dried and crushed plant material is often applied. As alkaloids usually possess amino and other functional groups that could react with solvents and additives, as well as with CO2 or oxygen from air, the formation of artefacts is prevented as far as possible by the choice of appropriate conditions. In general, artefacts can be recognized by comparing the analytical data of the isolated fractions and the original sample. Therefore, some of the original sample should always be kept as reference material (Hesse, 2000).
Materials and Reagents
- Cotton wool (e.g., Thomas Scientific, catalog number: 2904W25 )
- Sea sand (cristobalite, Brenntag Schweizerhall AG) (e.g., Grogg Chemie, catalog number: G890 )
- Round bottom test tubes, ca. 100 x 16 mm (Assistant, catalog number: 42775045 )
- TLC plates (TLC Silica gel 60 F254 on aluminum 20 x 20 cm) (EMD Millipore, catalog number: 100390 )
- Glass capillaries (MARCHEREY-NAGEL, catalog number: 814022 )
- 2 ml 9 mm ScreVial, clear glass 12 x 32 mm (Infochroma, catalog number: G004-HP-H )
- 9 mm screw cap with 1 mm pigment-free ms-pure PTFE/Silicone/PTFE-Septum (Infochroma, catalog number: G004-HP-CB-FKSKFK10 )
- 1.5 ml Eppendorf tubes (Eppendorf, catalog number: 0030120086 )
- Plant material: Capparis decidua root bark (collected in Sahiwal, Pakistan and identified by M. Waris, Department of Cholistan Institute of Desert Studies, The Islamia University of Bahawalpur, Pakistan)
- MeOH (methanol, technical grade, distilled prior to use) (Thommen Furler, catalog number: 203-VL54TE )
- SiO2 (Merck silica gel 60 [40-63 μm]) (EMD Millipore, catalog number: 109385 )
- Ammonium hydroxide (NH3, aqueous ammonia solution 25%, puriss p.a.) (Honeywell, catalog number: 30501 )
- Chloroform (CHCl3, stabilized with EtOH) (Scharlab, catalog number: CL02181000 )
- Dichloromethane (CH2Cl2, technical grade, distilled prior to use) (Thommen-Furler, catalog number: 739-VL54TE )
- Diethylether (Et2O, puriss. p.a., stabilized with BTH, distilled from NaOH prior to use) (Honeywell, catalog number: 32203 )
Notes: - Pure ether is sensitive towards oxidation and can form explosive peroxides upon prolonged exposure to air and light. It therefore is usually stabilized with the antioxidant butylhydroxytoluene (BHT), which is removed upon distillation. Thus, distilled ether is again prone to form epoxides, and it is necessary to test your distilled ether for peroxides prior to its use if the solvent was exposed to air and light for several days.
- To test for peroxides use: Quantofix® peroxides test sticks (Sigma-Aldrich, catalog number: Z101680 ).
- Methylamine solution (MeNH2, 40 wt. %) (Sigma-Aldrich, catalog number: 426466 )
- Hydrochloric acid (HCl, 37%) (for analysis, Merck) (EMD Millipore, catalog number: 100317 )
- Acetonitrile (MeCN, LC-MS Ultra CHROMASOLV®) (Honeywell, catalog number: 14261 )
- Formic acid (HCO2H, 99%, ULC/MS) (Biosolve, catalog number: 069141 )
- 2-propanol (LC-MS Ultra CHROMASOLV®) (Honeywell, catalog number: 650447 )
- Sodium hydroxide solution (NaOH for HPCE, 0.1 N in H2O) (e.g., MSP KOFEL, catalog number: 5062-8575 )
- H2PtCl6
- KI
- Ce(SO4)2
- H2SO4
- Schlittler reagent (see Recipes)
- Ce(SO4)2 solution (see Recipes)
Equipment
- Sharp axe (e.g., TRANSA, catalog number: 040929-001001 )
- Capped fermentation tank (50 L, e.g., Braupartner, catalog number: 141-0 )
- Filter strainer cloth (75 x 75 cm, e.g., Erwin Müller, catalog number: i10014278 )
- Stainless steel spatula (e.g., Thomas Scientific, catalog number: 1232X12 )
- Rotary evaporator (Büchi Rotavapor R-134 with Büchi Waterbath B-481) (Büchi, Flawil, Switzerland)
- Glass funnels (e.g., Duran, catalog number: 21 351 23 )
- Round bottom flasks of several sizes (5 ml, 10 ml, 25 ml, 50 ml, 250 ml, 1 L) (e.g., Schott, Germany)
- Steel spring clips (e.g., Thomas Scientific)
NS 14.5 (e.g., Thomas Scientific, catalog number: 1178Z55 )
NS 29 (e.g., Thomas Scientific, catalog number: 1178Z57 ) - Magnetic stir bars (e.g., Thomas Scientific, catalog number: 8608S78 )
- Magnetic stirrer (Heidolph Instrument, catalog number: 505-30080-00 )
- Glass bottle 1 L (e.g., Thomas Scientific, catalog number: 1228R95 )
- Glass chromatography columns of different length and diameters with stopcock at the bottom and a ground socket joint at the top to fit a solvent reservoir (e.g., Neubert Glas, catalog number: 1196-29-20400 )
- Glass beaker of several sizes (e.g., Duran, Germany)
- A piece of vacuum hose (e.g., Thomas Scientific, catalog number: 9544T15 )
- Solvent reservoirs: glass round bottom flasks of several sizes with oppositely attached ground cone and ground socket joints (e.g., Thomas Scientific, catalog number: 1197C03 )
- Adaptors:
Ground cone joint NS 14.5 with hose barbe to tubing, bent (e.g., Thomas Scientific, catalog number: 1195C09 )
Ground cone joint NS 29 with hose barbe to tubing, bent (e.g., LabMarket, catalog number: 1681029 ) - Hand pump (VWR, catalog number: 612-9952 )
- 125 ml Erlenmeyer flasks (e.g., Thomas Scientific, catalog number: 4907F23 )
- Test tube racks (e.g., Thomas Scientific)
- TLC chamber (screw cap glass jar, 250 ml)
- UV lamp (254 nm), e.g., UV lamp 4, dual (Camag, model: 022.9160 )
- High vacuum pump (Alcatel Pascal 2015 SD) (e.g., Ideal Vaccum Products, catalog number: P102310 )
- CortecsTM UPLC® C18+, 1.6 μm, 2.1 x 150 mm (Waters, catalog number: 186007117 ), equipped with a CortecsTM UPLC® C18+ 1.6 μm, 2.1 x 5 mm VanGuardTM precolumn (Waters, catalog number: 186008713 )
- AquityTM Ultra Performance LC (Waters, Milford MA, USA)
- Bruker maXis Q-Tof HR-MS (Bruker Daltonics, Bremen, Germany)
- Eppendorf refrigerated microcentrifuge (Eppendorf, model: 5417R )
- MilliQ gradient apparatus (deionized water, for HPLC) (EMD Millipore, catalog number: Z00Q0V0WW )
Procedure
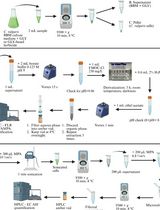
- Extraction of the root bark
- Dry the root bark in the shade for one week at 30-35 °C.
Note: If this is not your regular ambient temperature you may use a drying oven. - Shred the bark finely with a sharp axe and extract it with MeOH (1:1, w/v) in a capped fermentation tank for one week. Stir the mixture several times a day. If possible: stir your extraction mixture permanently with a mechanical stirrer.
Note: In our case, the extraction was performed under regular laboratory condition at ambient temperature (30-35 °C in Pakistan). - Filter off the solid through a tightly woven cloth and evaporate the MeOH on the rotary evaporator at 30 °C and 70 mbar for several hours, upon which the extract becomes highly viscous. Since it is not possible to completely remove the MeOH, stop with the evaporation of the solvent when no longer the formation of bubbles in the extract can be observed.
- Store the almost black, viscous extract at 4 °C until it is used.
- Dry the root bark in the shade for one week at 30-35 °C.
- Chromatographic separation of the alkaloids: general procedures
The chromatographic separation of the codonocarpine type alkaloids on silica gel (SiO2) involves at least four chromatographic separations that follow all the main principles described below:- Adsorption of the sample on SiO2
The crude extract and the alkaloid fractions are poorly soluble in small amounts of MeOH and even less soluble in the eluent that is used for the chromatography. It is, however, very important to apply the extract onto the chromatography column within the smallest possible volume. Therefore, the mixtures were adsorbed on SiO2 before they were brought onto the column. - Transfer the sample into a round bottom flask and add MeOH (1:4, w/v). Take a large round bottom flask (we used a 250 ml flask for 10.8 g of extract) to ensure that the glass surface is large enough to secure later a rapid and efficient evaporation of the solvent. Add a magnetic stir bar and stir vigorously until you obtain a uniform (still heterogeneous) mixture.
- While still stirring, add SiO2 (sample/SiO2 1:2, w/w), and continue with stirring for another 5 min.
- Remove the stir bar and then remove the MeOH on the rotary evaporator (30 °C, 160 mbar) until you obtain a bright brown powder.
- Preparation of the eluent mixture and the column
- Mix the eluent components in the given ratio (Tables 1-5) in a clean glass bottle. It is crucial to distil the solvents prior to use.
- Prepare a column as shown in Figure 2.
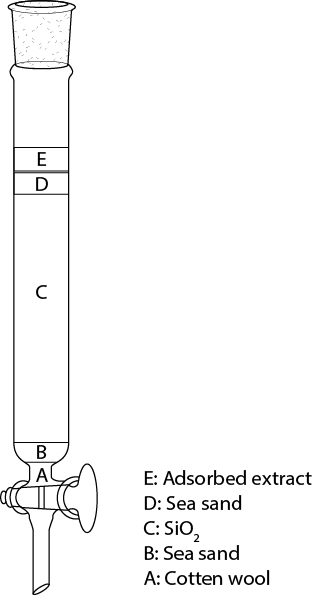
Figure 2. Packing of a chromatographic column. The columns were stacked layers of cotton wool (A), sea sand (B), SiO2 (C), additional sea sand (D) and the adsorbed sample on SiO2 (E).- Fill the column with approximately 5 cm of the eluent and seal it loosely and free of air at the bottom with some cotton wool (Figure 2A) to prevent the SiO2 from flowing out.
- Add 1 cm of sea sand (Figure 2B) so that you obtain a plane surface.
- In a beaker, prepare a slightly viscous, bubble-free suspension of SiO2 in the eluent. Add eluent until the SiO2 is well covered and stir with a spatula until all air has escaped.
- Fill the SiO2 suspension through a glass funnel carefully, not disturbing the surface of the sea sand, into your glass column to obtain a loose column of SiO2 (Figure 2C).
- Open the stopcock and let the eluent escape until the solvent surface exactly reaches the surface of the SiO2. Close the stopcock.
- Compact the SiO2 column by tapping against the glass column with a piece of vacuum hose. Upon tapping you can observe that the SiO2 level drops. Open the stopcock from time to time to drain the solvent again to the SiO2 surface. Continue compacting until the SiO2 surface no longer sinks.
- Add approximately 5 cm of solvent and trickle approximately 2 cm of sea sand (Figure 2D) through the solvent onto the surface of your column to protect the column against mechanical perturbations. Drain the solvent until its surface reaches the surface of the sea sand.
- Apply the brown powder of your adsorbed substance onto the column (Figure 2E), add carefully 2 cm of eluent, then drain the eluent carefully until it reaches the surface of your SiO2 with the adsorbed extract. Repeat the addition and draining of solvent three times.
- Carefully fill up the column with the eluent, connect the solvent reservoir, fix it with a clamp, and fill the reservoir with eluent.
- Fill the column with approximately 5 cm of the eluent and seal it loosely and free of air at the bottom with some cotton wool (Figure 2A) to prevent the SiO2 from flowing out.
- Mix the eluent components in the given ratio (Tables 1-5) in a clean glass bottle. It is crucial to distil the solvents prior to use.
- Chromatographic separation
- To start your separation, open the stopcock and adjust the flow of the eluent to an appropriate rate. Depending on the size of the column, this shall be approx. 12 ml min-1 (coarse fractionation) or approx. 4 ml min-1 (fine fractionation). If the flow rate is too low with a completely opened stopcock, apply slight but constant pressure with a manual pump.
- Collect fractions of reasonable volumes. The volumes to choose depend on the size of the column and the intended purpose of the chromatography. For a preliminary, rough separation of the original crude extract on a large column, 100 ml fractions, collected in Erlenmeyer flasks, are appropriate. For fine separations on smaller columns, preferential fractions of approx. 10 ml are collected in round bottom test tubes. Since the alkaloids of interest are readily recognizable as yellow bands on the column, the clearly colourless pre-fractions are usually collected in Erlenmeyer flasks.
- Fraction control by TLC
- Transfer a small amount of your fraction with a glass capillary on a TLC plate and let the spot dry. You can re-use the same capillary several times when you clean it in-between with some MeOH.
- Place the TLC plate in a TLC chamber (Figure 3) with 3-5 mm of one of the following eluents:
- CH2Cl2/MeOH/NH3 40:13:1: to test if your fraction contains any spermidine alkaloids at all. The typical Rf value for the spermidine alkaloids is shown in Figure 3.
- MeOH/H2O/NH3 15:2:1: to test which spermidine alkaloid is in your fraction. This eluent was developed by Kyburz (1985) and was the only one that was found to distinguish between the alkaloids of the different masses. The typical Rf values for the spermidine alkaloids are given in Figure 3.
- Let the TLC develop till the solvent front reaches almost the top of the plate, remove the TLC plate from the chamber, and let it dry until the plate no longer smells of NH3.
- Check for UV-active spots under a UV lamp (254 nm) and mark them with a pencil.
- Stain your TLC plate with Schlittler reagent and dry it. The spermidine alkaloids give red/violet spots. When the dry Schlittler-active spots are stained subsequently with Ce(SO4)2 solution, the colour of the spots deepens. According to our experience, the detection by UV is the most sensitive method. Combined with the staining with Schlittler reagent, the spots for the alkaloids of interest are very specifically and clearly recognized. The treatment with Ce(SO4)2 is helpful but not necessary.

Figure 3. TLC chamber and the schematic TLCs with the two most useful eluents for the detection of spermidine alkaloids. For the detection of the alkaloid fractions in the preliminary rough chromatography use the eluent system CH2Cl2/MeOH/NH3 40:13:1 (gives no separation of the individual alkaloids, left), and for fine separation of the alkaloids use the eluent system MeOH/H2O/NH3 15:2:1 (separation of the individual alkaloids, right). However, codonocarpine (2) and isocodonocarpine (3) remain indistinguishable by TLC. - Combination of the fractions and evaporation of the solvent
- Combine the fractions according to their TLCs. Pay attention not to pool pure alkaloid fractions with fractions that contain alkaloid mixtures. Mind that the alkaloids are chromatographing quite closely, and a lowly abundant alkaloid next to a major component may not be easily recognized by TLC! Very small amounts of an alkaloid are only detected by UV light but not with the Schlittler staining reagent.
- Evaporate the solvent on the rotary evaporator at 30 °C (pressure depending on your eluent), and remove residual solvent at high vacuum (23 °C, 4 x 10-4 mbar).
Note: To determine the net mass of your sample subtract the tare mass (empty flask) from the gross mass (sample + flask). - Control your dried samples by HPLC-MS (see Data analysis). Check their final alkaloid composition with the extracted ion chromatograms (EIC) m/z 436.2231 (1), m/z 466.2237 (2/3) and m/z 496.2442 (4).
- Adsorption of the sample on SiO2
- Chromatographic separation of the four major alkaloids in detail
Notes: This section contains a detailed description of what was done in our laboratory. For an improved separation, we recommend the following steps:
1. Start with a rough alkaloid isolation as done in column B (Table 3).
2. Separate the alkaloids 1 and 2/3 from alkaloid 4 as described for column B1 (Table 4).
3. Combine all fractions containing 1 and 2/3 to one sample and chromatograph this as described in column A1.2 to obtain pure 1 and 2/3 (Table 2).
4. Wash the fraction containing 4 with MeOH as described for B1.4.1.
The four alkaloids cadabicine (1, 435 Da), codonocarpine (2, 465 Da), isocodonocarpine (3, 465 Da) and capparidisinine (4, 495 Da) have very similar chromatographic behaviours. For this reason, we obtained at the beginning of our project many mixed fractions upon preliminary chromatographies of the crude original extract as well as of partially separated fractions. For the isolation of 1 and 2/3 we started with an alkaloid mixture (A1, 576 mg) that was obtained by the combination of several of these fractions. A pre-purified alkaloid fraction that could be used likewise is fraction B1, employed below for the isolation of capparidisine (4).- Path A: Isolation of cadabicine (1) and codonocarpine (2)/isocodonocarpine (3)
The separation path A is schematically shown in Figure 4. The alkaloid-containing fractions of various preliminary chromatographies (A) were combined in A1. The chromatographic separation of A1 led to four major fractions. Three of them contained mixtures of 1 and 2/3 in diverse ratios (A1.1 mainly 1, A1.2 comparable amounts of 1 and 2/3, and A1.3 mainly 2/3). The fourth fraction (A1.4) contained mainly alkaloid 4 but also some 2/3. As none of the obtained fractions contained a pure alkaloid, the samples A1.1 and A1.3 were kept as reserve material and the largest fraction (A1.2) was further separated on SiO2 to obtain the alkaloids 1 (A1.2.1) and 2/3 (A1.2.3) in high purity (> 98%).
Unfortunately, fraction A1.4 contained not enough material for the final isolation of pure 4.
The chromatic details and results are summarized in the Tables 1 and 2. In fractions of alkaloid mixtures, the major alkaloid is underlined.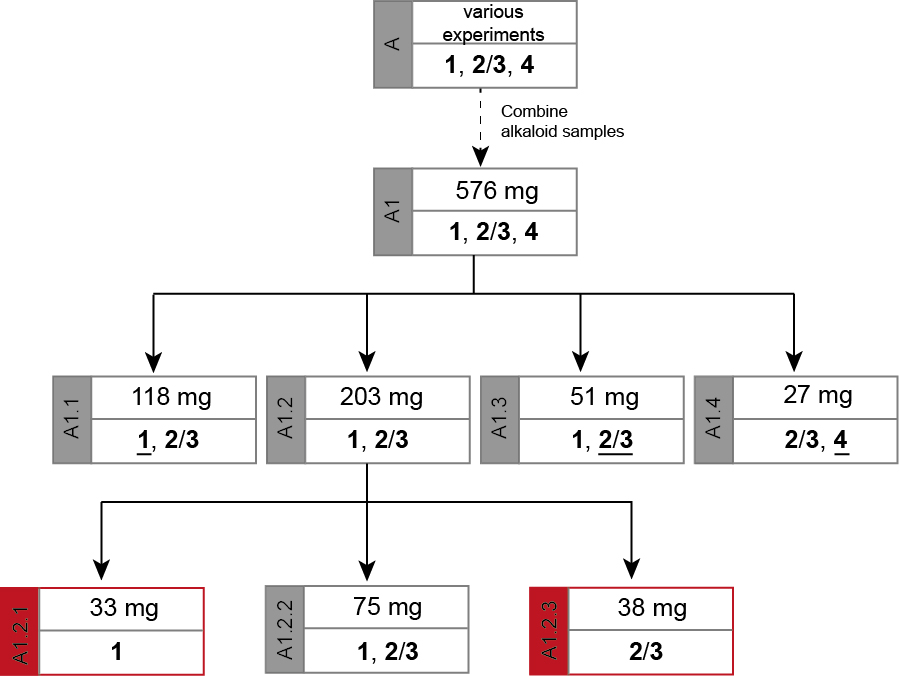
Figure 4. Separation path A. Overview over the chromatographic steps from an alkaloid mixture (A1) to the pure sample of cadabicine (1, A1.2.1) and the mixed sample of codonocarpine and isocodonocarpine (2/3, A1.2.3) that were used to elucidate their structures. The major alkaloid in a mixed fraction is underlined.
Table 1. Chromatographic separation of A1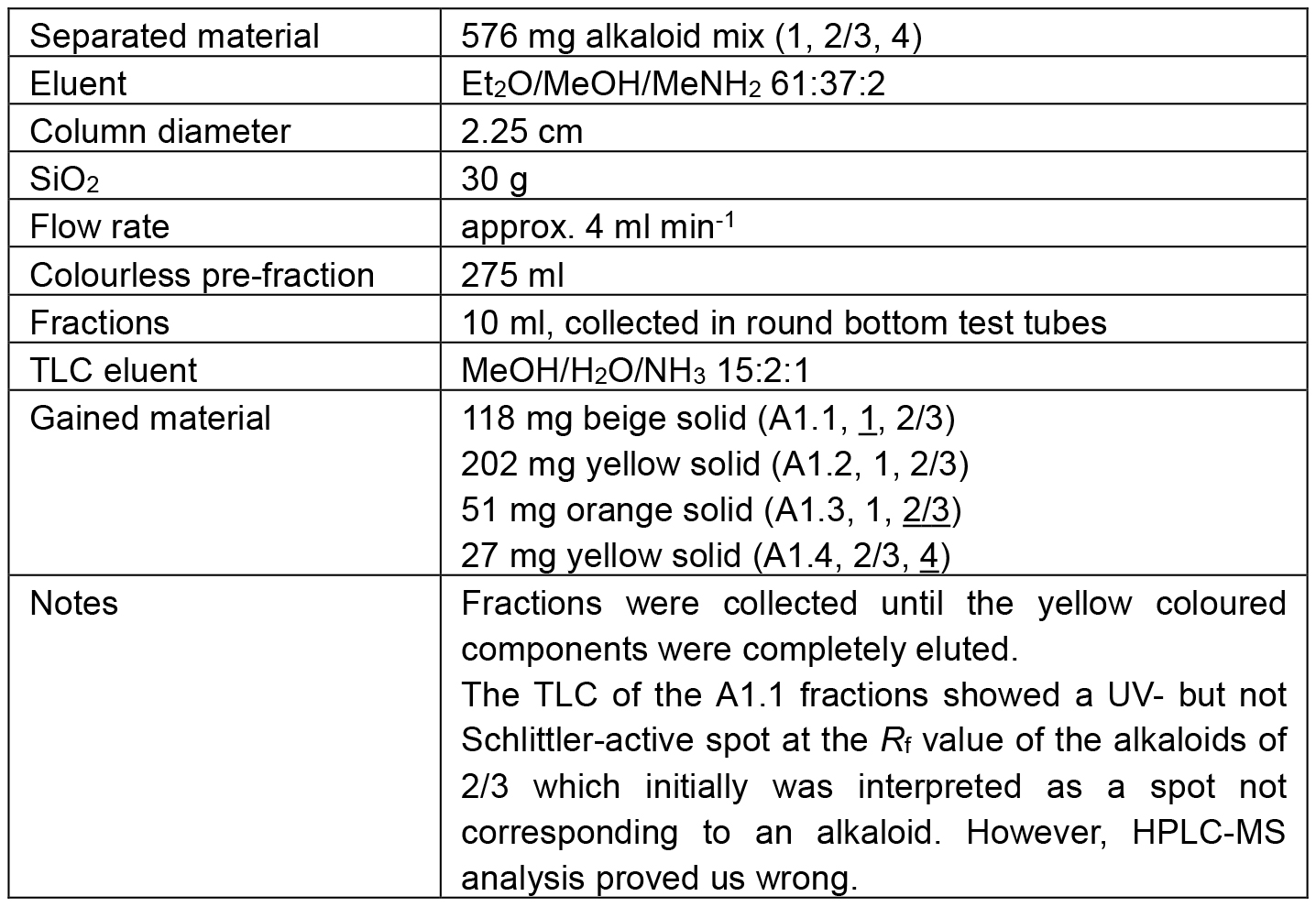
Table 2. Chromatographic separation of A1.2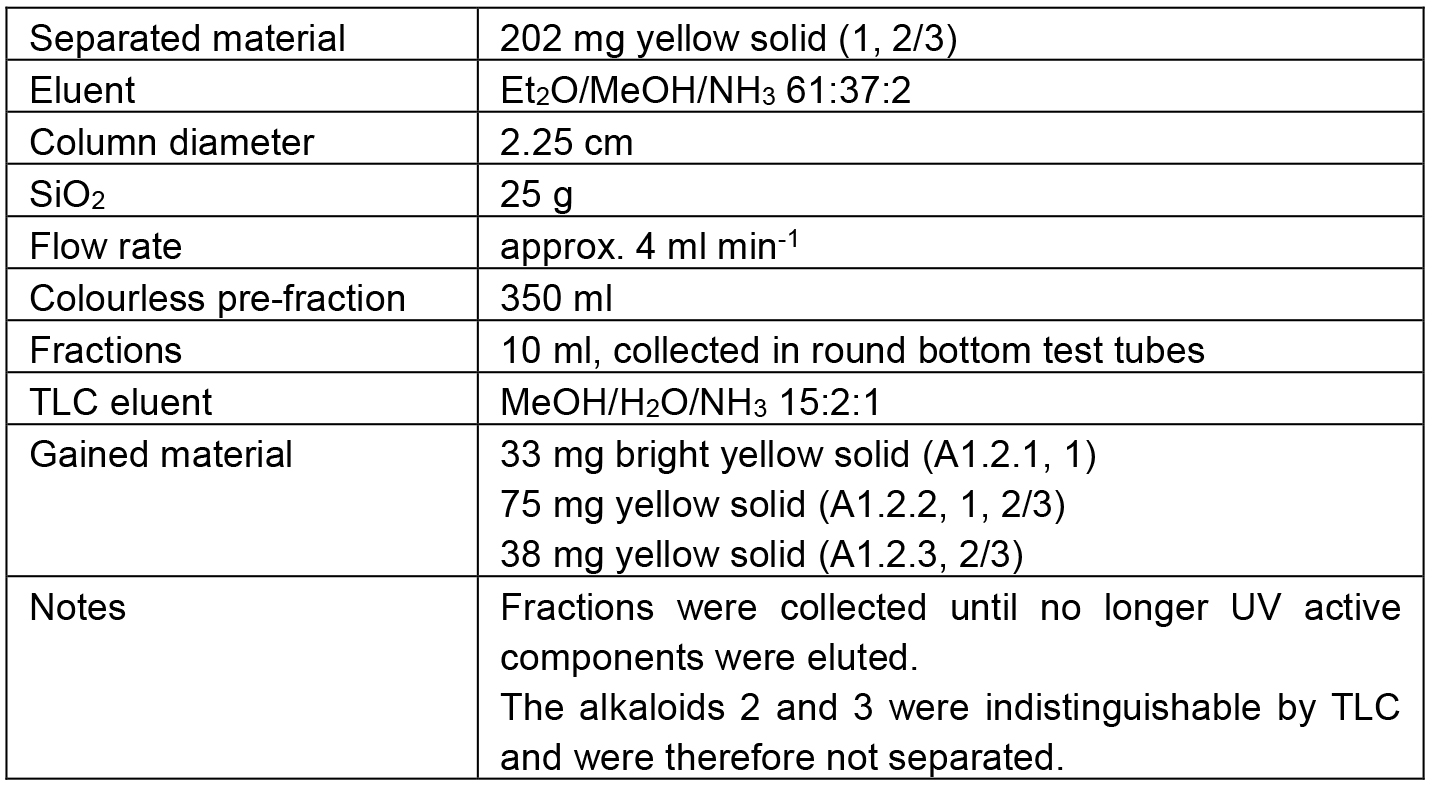
- Path B: Isolation of capparidisinine (4)
The separation path B is schematically shown in Figure 5. The crude methanolic extract (B) was roughly fractionated on SiO2 to separate the alkaloid-containing fractions (B1) from the further extracted material. The alkaloid mixture B1 was chromatographed to obtain three fractions containing different ratios of 1 and 2/3 (B1.1-B1.3) and one fraction containing only one alkaloid (4). Alkaloid 4 in fraction B1.4 however, was contaminated with some unidentified Schlittler-inactive compounds (detected by 1H-NMR). An additional chromatographic step (B1.4.1) could not separate the alkaloid 4 from its contaminates. Capparidisinine (4, B1.4.1.1) was finally gained by washing sample B1.4.1 with small amounts of MeOH.
The chromatic details and results are summarized in the Tables 3-5. In fractions of alkaloid mixtures, the major alkaloid is underlined.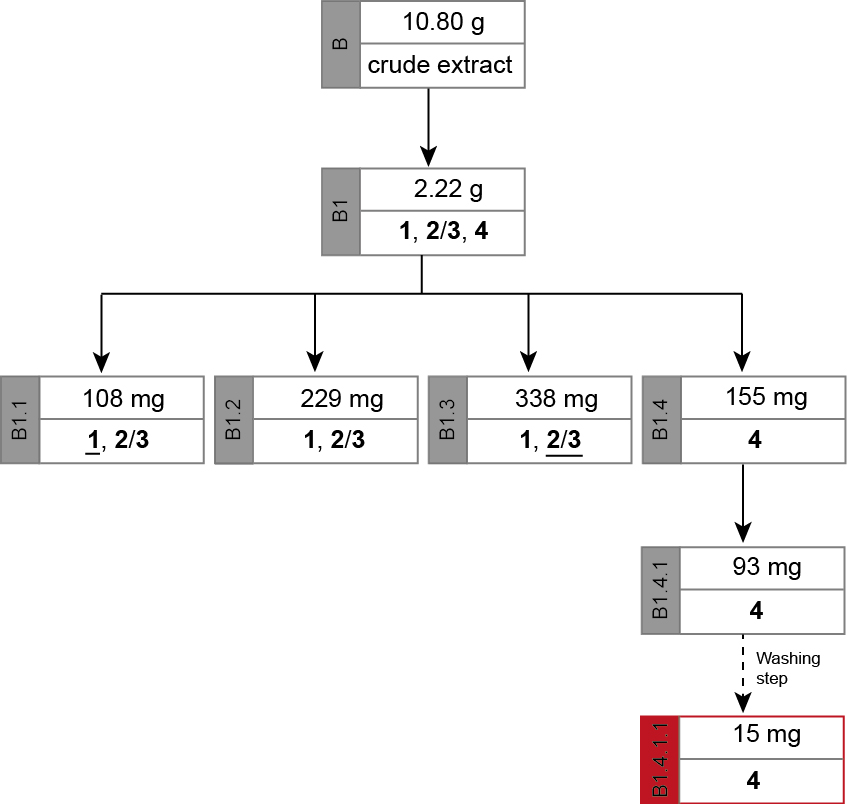
Figure 5. Separation path B. Overview over the chromatographic steps from the crude extract (B) to alkaloid 4 (B1.4.1.) inclusive the washing step to obtain the pure form of capparidisinine (4, B1.4.1.1) from which its structure was elucidated. The major alkaloid in a mixed fraction is underlined.
Table 3. Chromatographic separation of B
Table 4. Chromatographic separation of B1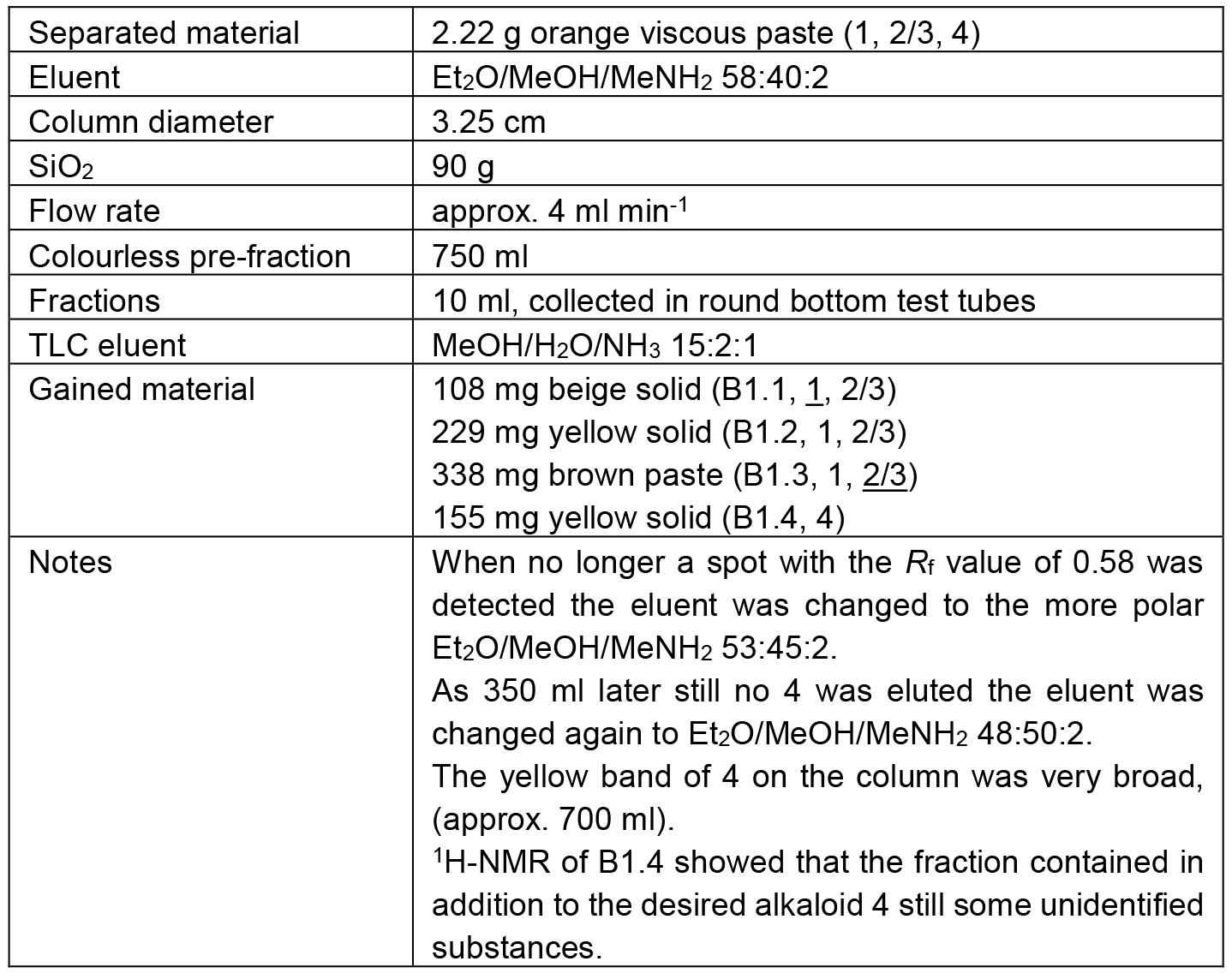
Table 5. Chromatographic separation of B1.4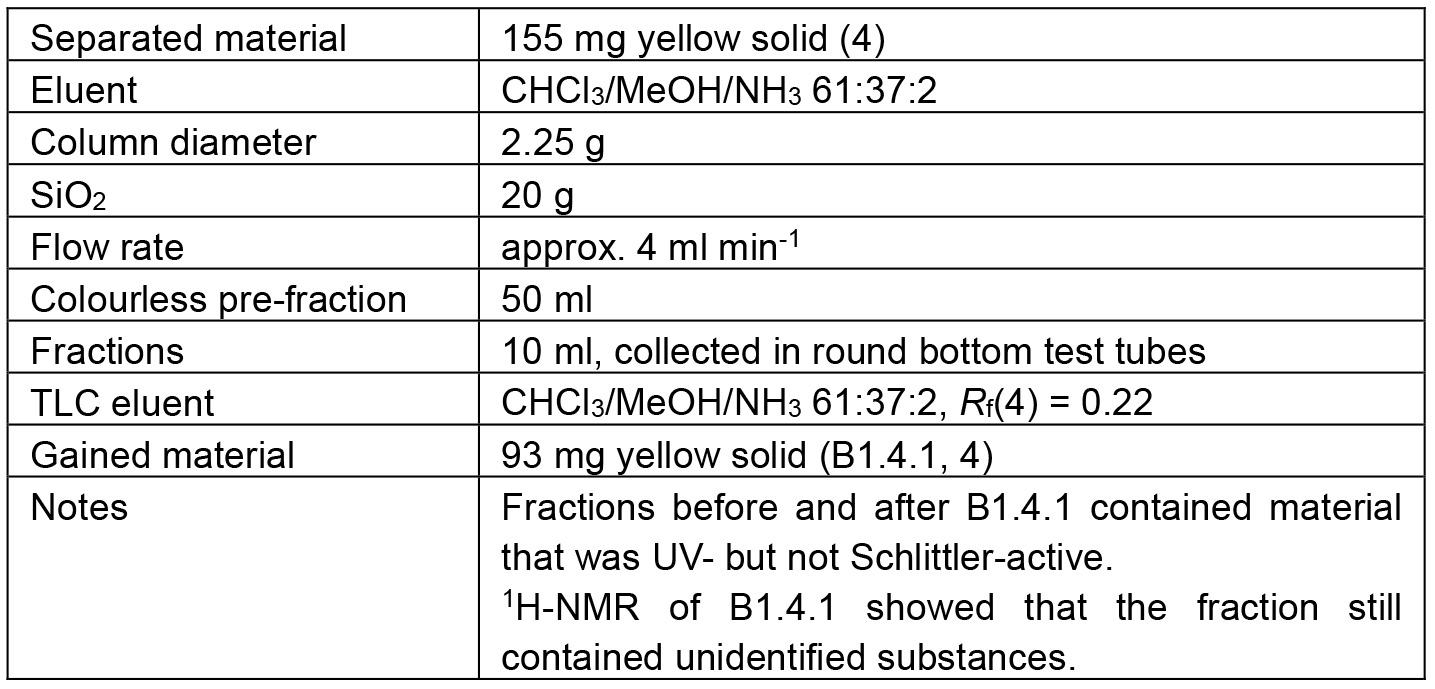
- Further purification of B1.4.1
The yellow solid of B1.4.1 (77 mg) was placed in a 1.5 ml Eppendorf tube, and MeOH (0.3 ml) was added. The cloudy mixture was shaken carefully and then centrifuged (1 min, 20,817 x g). The yellow solution was carefully separated from the remaining solid with a pipette. The solid was washed three more times with MeOH (0.2 ml each time). The remaining solid was transferred to a round bottom flask, and dried in vacuo (30 °C, 160 mbar then 23 °C, 0.2 mbar) to deliver a bright yellow solid B1.4.1.1 (4, 15 mg).
- Path A: Isolation of cadabicine (1) and codonocarpine (2)/isocodonocarpine (3)
- Conversion to the hydrochloride salts
- Suspend the alkaloid in MeOH.
- Add dropwise 1.5 eq of aq. HCl (0.1 or 1.0 N) to the cloudy solution. The solution becomes clear.
- Evaporate the solvent in vacuo (30 °C, 160 mbar then 23 °C, 4 x 10-4 mbar).
- Suspend the alkaloid in MeOH.
Data analysis
- HPLC-ESI-MS
- The alkaloid fractions were analysed by HPLC-ESI-MS. The samples were dissolved in MeOH + 0.1% HCO2H at 0.5 mg ml-1 for the crude extract and 0.1 mg ml-1 for the purified alkaloids. The samples were injected at a volume of 1 μl and chromatographed at a flow rate of 0.3 ml min-1 on a RP C18+ column with the solvents A (H2O + 0.1% HCO2H) and B (MeCN + 0.1% HCO2H) at 25 °C. The gradient started isocratic at 10% B for 5 min and raised then to 25% B within 4 min. The column was washed with 100% B for 2 min and then equilibrated at 5% B for 6 min.
- The connected Q-Tof MS was equipped with an ESI source. The MS was recorded in positive ionisation mode from 50-800 m/z under the following conditions: HV end plate offset 500 V, HV capillary 3,000 V, nebulizer gas (N2) 2.0 bar, dry gas (N2) 10.0 bar, and dry temperature 200 °C. The mass spectrometer was calibrated for mass accuracy with 2 mM sodium formate (980 μl H2O/2-propanol 1:1, 20 μl 0.1 N NaOH and 1 μl HCO2H), the relative mass error being typically lower than 2 ppm (externally).
- The MS/MS spectra were recorded with 35.00 eV collision energy and 4.00 isolation width.
- The chromatograms and spectra were evaluated with DataAnalysis. The extracted ion chromatograms (EIC) were educed based on the exact mass ± 0.02 m/z.
While cadabicine (1) and capparidisinine (4) can be distinguished with HR-MS, this is not the case for codonocarpine (2) and isocodonocarpine (3). We found that codonocarpine (2) has a slightly smaller retention time (4.2 min) than isocodonocarpine (3, 4.3 min). The MS/MS spectra for (2) and (3) are virtually the same. They differ only by one low-intense signal at m/z 218.08058 that is unique for codonocarpine (2). However, be very careful; both compounds show a signal at m/z 218.11713.
Note: For a detailed discussion of the MS/MS spectra and the interpretation of the NMR spectra check ‘A new sight on the Codonocarpine Type Alkaloids of Capparis decidua’ (Forster et al., 2016).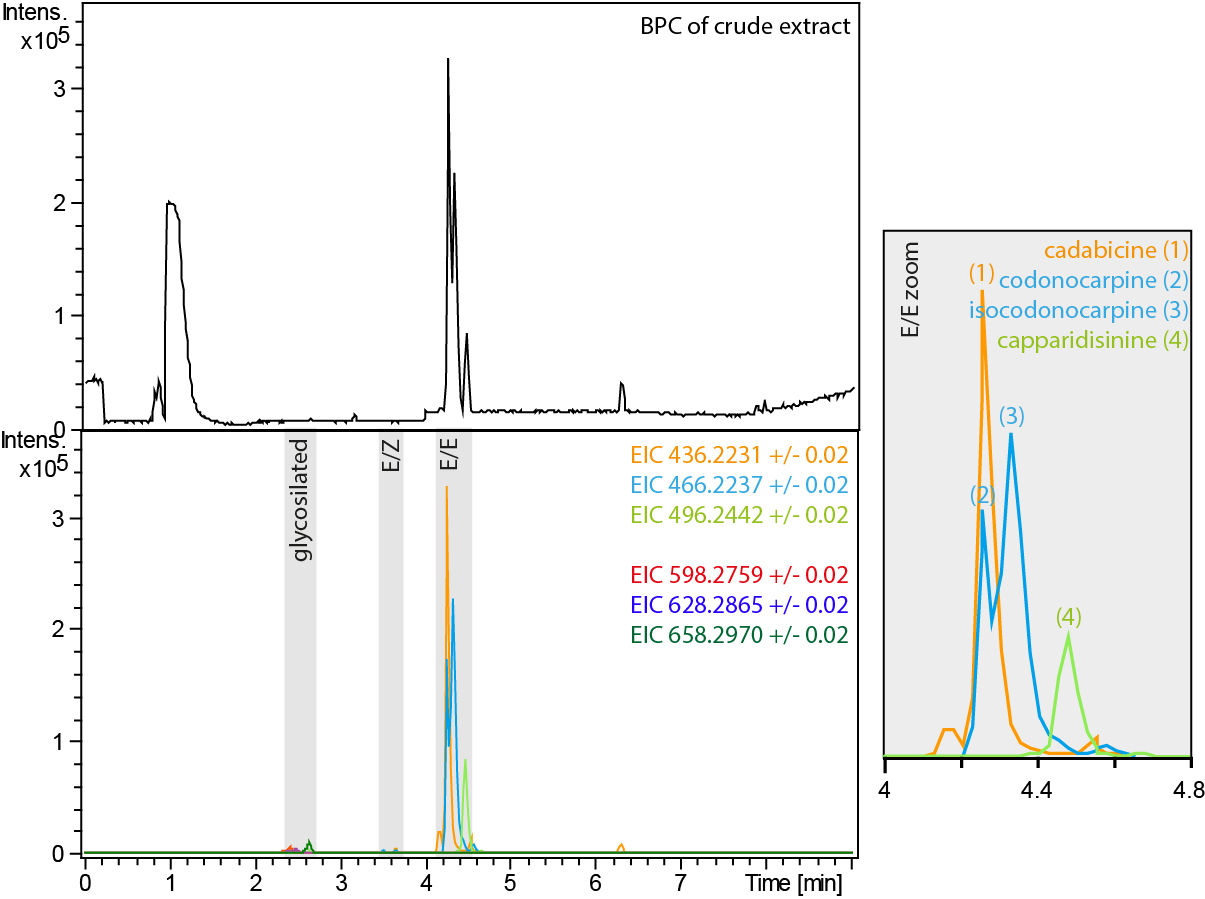
Figure 6. BPC of the crude extract and the EICs of the codoncarpine type alkaloid related masses. The crude extract contains three different forms of alkaloid derivatives. The four major alkaloids cadabicine (1), codonocarpine (2), isocodonocarpine (3) and capparidisinine (4), all (E/E) derivatives, can be identified by their exact mass and retention time. - The alkaloid fractions were analysed by HPLC-ESI-MS. The samples were dissolved in MeOH + 0.1% HCO2H at 0.5 mg ml-1 for the crude extract and 0.1 mg ml-1 for the purified alkaloids. The samples were injected at a volume of 1 μl and chromatographed at a flow rate of 0.3 ml min-1 on a RP C18+ column with the solvents A (H2O + 0.1% HCO2H) and B (MeCN + 0.1% HCO2H) at 25 °C. The gradient started isocratic at 10% B for 5 min and raised then to 25% B within 4 min. The column was washed with 100% B for 2 min and then equilibrated at 5% B for 6 min.
Notes
We found that the most stable form to store the codonocarpine type alkaloids is in their solid forms. The fractions were therefore always dried on the rotary evaporator immediately after chromatography.
Recipes
- Schlittler reagent (Schlittler and Hohl, 1952)
The mixture of H2PtCl6 (1.0 g in H2O [6 ml]), KI (22.5 g in H2O [225 ml]), aq. HCl solution (1 N, 20 ml) was diluted with deionized H2O to a total volume of 1,000 ml
Colouring,
Amines: brown in diverse shades (yellow-red-blue-grey)
Amides: bright yellow, followed by Ce(SO4)2: brown - Ce(SO4)2 solution
A solution of Ce(SO4)2 (10.0 g) in conc. H2SO4 (55 ml) was diluted with deionized H2O to at total volume of 1,000 ml
Colouring,
Amines: (together with Schlittler reagent) brown
Amides: (together with Schlittler reagent) intensifies the staining colour of the Schlittler reagent
Acknowledgments
We are grateful to PD Dr. L. Bigler for his great support in the acquisition of the MS and MS/MS data and we thank the Higher Education Commission of Pakistan (HEC) (1-8/HEC/HRD/2013/2582, PIN:IRSIP 23 Ps 48) for funding a six-month research visit of A. Ghaffar to the University of Zurich.
References
- Ahmad, V. U., Arif, S., Amber, A. R. and Fizza, K. (1987). Capparisinine, a new alkaloid from Capparis decidua. Liebigs Ann der Chemie 2: 161-162.
- Ahmad, V. U., Arif, S., Amber, A. R. and Nasir, M. A. (1986). A new alkaloid from root bark of Capparis decidua. Zeitschrift für Naturforschung B 41(8): 1033-1035.
- Ahmad, V. U., Arif, S. Amber, A. R., Usmanghani, K. and Miana, C. A. (1985). A new spermidine alkaloid from Capparis decidua. Heterocycles 23: 3015-3020.
- Ahmad, V. U., Ismail, N. and Amber, A. R. (1989). Isocodonocarpine from Capparis decidua. Phytochemistry 28: 2493-2495.
- Arif, S. (1986). Studies on alkaloids of Capparis decidua. Dissertation.
- Bienz, S., Detterbeck, R., Ensch, C., Guggisberg, A., Häusermann, U., Meisterhans, C., Wendt, B., Werner, C. and Hesse, M. (2002). The Alkaloids, Vol 58. Academic Press pp: 83-338.
- Forster, Y., Ghaffar, A. and Bienz, S. (2016). A new view on the codonocarpine type alkaloids of Capparis decidua. Phytochemistry 128: 50-59.
- Hesse, M. (2000). Alkaloide - Fluch oder Segen der Natur? Verlag Helvetica Chimica Acta.
- Kyburz, R. (1985). Alkaloide aus Aristotelia peduncularis (Labill.) Hook. F. und aus Capparis decidua (Forsk.) Edgew. Dissertation.
- Mohammed, M. S., Khalid, H. S., Muddathir, A. E., K. Tahir, El, Khan, A. A., Algadir, H. A., Ahmed Osman, J. W. and Siddiqui, N. A. (2015). Effect of some plants’ extracts used in Sudanese folkloric medicines on carrageenan-induced inflammation. Pak J Pharm Sci 28(1): 159-165.
- Schlittler, E. and Hohl, J. (1952). Über die Alkaloide aus Strychnos melinoniana Baillon. Helv Cimica Acta 35: 29-45.
- Singh, D. and Singh, R. K. (2011). Kair (Capparis decidua): A potential ethnobotanical weather predictor and livelihood security shrub of the arid zone of Rajasthan and Gujrat. Indian J Tradit Knowl 10: 146-155.
- Singh, P., Mishra, G., Srivastava, S., Jha, K. K. and Khosa, R. L. (2011). Traditional uses, phytochemistry and pharmacological properties of Capparis decidua: An overview. Der Pharm Lett 3: 71-82.
- Tlili, N., Elfalleh, W., Saadaoui, E., Khaldi, A., Triki, S. and Nasri, N. (2011). The caper (Capparis L.): Ethnopharmacology, phytochemical and pharmacological properties. Fitoterapia 82(2): 93-101.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Forster, Y., Ghaffar, A. and Bienz, S. (2017). Chromatographic Separation of the Codonocarpine Type Alkaloids from the Root Bark of Capparis decidua. Bio-protocol 7(4): e2144. DOI: 10.21769/BioProtoc.2144.
Category
Plant Science > Plant biochemistry > Other compound
Plant Science > Plant physiology > Metabolism
Biochemistry > Other compound > Alkaloid
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.