- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

P-body and Stress Granule Quantification in Caenorhabditis elegans


Published: Vol 7, Iss 2, Jan 20, 2017 DOI: 10.21769/BioProtoc.2108 Views: 14212
Reviewed by: Jyotiska ChaudhuriLeonardo G. GuilgurPia Giovannelli
Original research article
The authors used this protocol in:

May 2015

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Eukaryotic cells contain various types of cytoplasmic, non-membrane bound ribonucleoprotein (RNP) granules that consist of non-translating mRNAs and a versatile set of associated proteins. One prominent type of RNP granules is Processing bodies (P bodies), which majorly harbors translationally inactive mRNAs and an array of proteins mediating mRNA degradation, translational repression and cellular mRNA transport (Sheth and Parker, 2003). Another type of RNP granules, the stress granules (SGs), majorly contain mRNAs associated with translation initiation factors and are formed upon stress-induced translational stalling (Kedersha et al., 2000 and 1999). Multiple evidence obtained from studies in unicellular organisms supports a model in which P bodies and SGs physically interact during cellular stress to direct mRNAs for transport, decay, temporal storage or reentry into translation (Anderson and Kedersha, 2008; Decker and Parker, 2012). The quantification, distribution and colocalization of P bodies and/or SGs are essential tools to study the composition of RNP granules and their contribution to fundamental cellular processes, such as stress response and translational regulation. In this protocol we describe a method to quantify P bodies and SGs in somatic tissues of the nematode Caenorhabditis elegans.
Keywords: Caenorhabditis elegansBackground
Thus far, most protocols to study P bodies and SGs were developed for yeast or human cell lines (Buchan et al., 2010). Little is known about the function of somatic RNP granules in multicellular organisms. The simple model organism C. elegans has been extensively used to study germline-specific P granules, which are distinct from P bodies and SGs, and important structures for germline development and function (Updike and Strome, 2010). Although the principles of the presented procedure can be applied to count germline-specific P granules, the protocol focusses on the quantification of somatic RNP granules. Several studies have identified a conserved function of somatic P bodies in the translational deregulation via miRNA pathways in C. elegans (Ding et al., 2005; Zhang et al., 2007). More recently, various tools were created to study the involvement of cytoplasmic RNP granules in cellular and organismal stress response, development and ageing in the nematode (Cornes et al., 2015; Huelgas-Morales et al., 2016; Rieckher et al., 2015; Rousakis et al., 2014; Sun et al., 2011; Table 1).
Such studies take advantage of the comparatively easy implementation of transgenesis methods in C. elegans that allow to constitutively express fluorescent fusion proteins (e.g., green fluorescent protein [GFP]), endogenously or in specific tissues (Rieckher et al., 2009). A collection of fosmids carrying gfp-tagged P body- and SG-specific genes can be obtained at the ‘C. elegans TransGeneome’ project, a genome-scale transgenic project for fluorescent- and affinity-tagged proteins for expression in the nematode (Sarov et al., 2012; Table 1). C. elegans is transparent, which allows for efficient application of fluorescence microscopy methods that are easily combined with differential interference contrast (DIC) microscopy to reveal fluorescent protein expression in an anatomical context. Mounting transgenic animals for P body and SG imaging is based on a previously described method using nanoparticles for immobilization (Kim et al., 2013), since commonly applied anesthetics in C. elegans can induce stress, resulting in increased RNP granule formation. Fluorescence-tagged P body or SG-intensity can be imaged by epifluorescence light microscopy (see Procedure C), while fluorescence intensity, a detailed count and size measurements of P bodies and SGs can be obtained via confocal laser scanning microscopy (see Procedure D).
Table 1. Tools available for transgenic expression of P body/SG-factors in C. elegans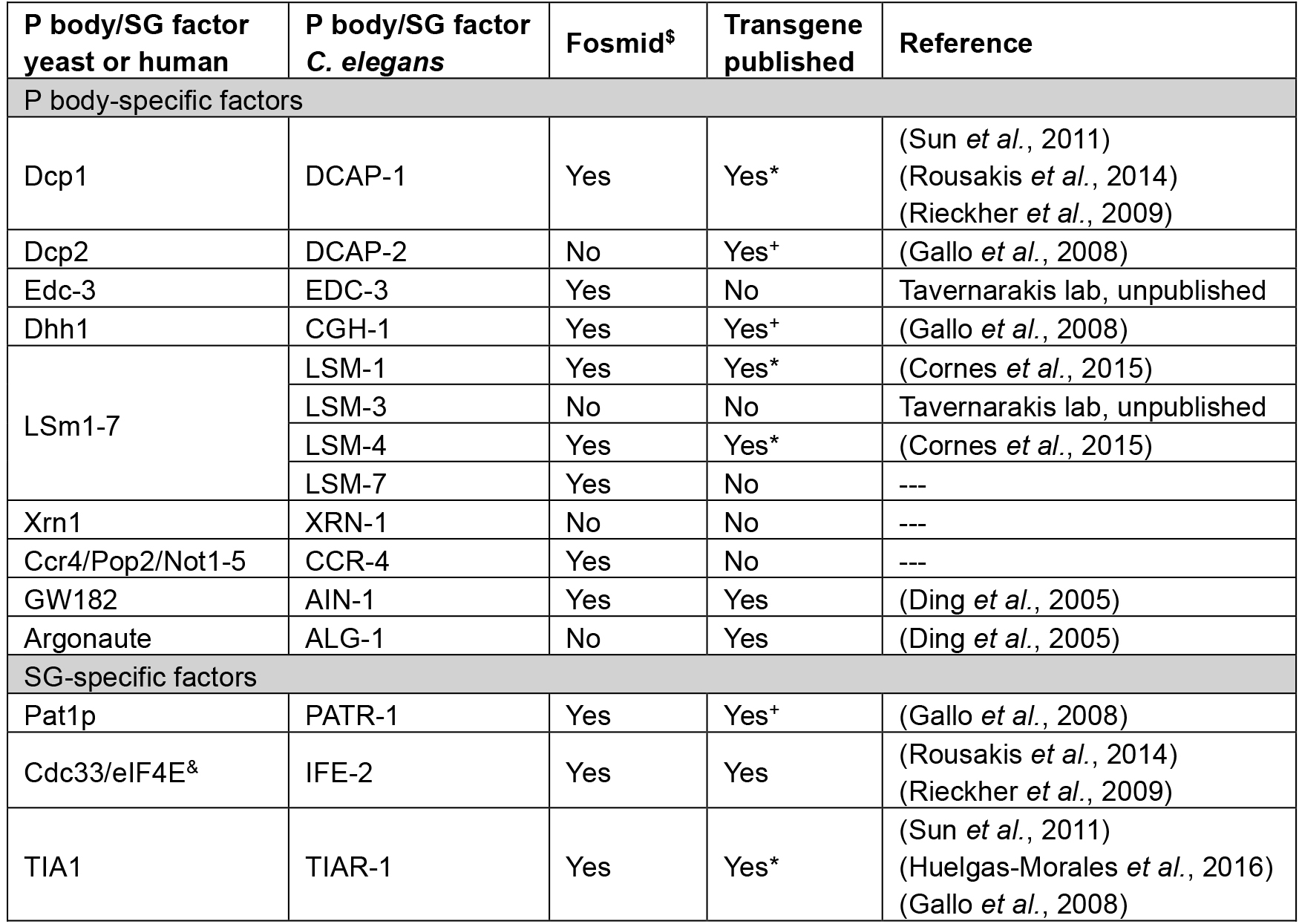
*available at CGC
+germline-specific promoter fusion
$C. elegans TransGeneome project (Sarov et al., 2012)
&can be found in both types of RNP granules
Materials and Reagents
- Sterile pipette tips
- Surgical disposable scalpel (Braun Medical, catalog number: 5518075 )
- Worm Pick with platinum wire (Genesee Scientific, catalog numbers: 59-30P6 )
- Pre-flattened tip (Genesee Scientific, catalog numbers: 59-AWP )
- Microscope slides (76 x 26 mm) (Carl Roth, catalog number: 0656.1 )
- Cover slips (24 x 24 mm) (Carl Roth, catalog number: H875.2 )
- Tape (~1 mm thickness)
- Greiner Petri dishes (60 x 15 mm) (Greiner Bio One, catalog number: 628161 )
- C. elegans strains (see Table 1 for available transgenes)
- Escherichia coli OP50 strain (obtained from the Caenorhabditis Genetics Center)
- Nail polish
- Polystyrene beads (Polybead, 2.5% by volume, 0.1 µm diameter)
- Potassium dihydrogen phosphate (KH2PO4) (Carl Roth, catalog number: P018.1 )
- Di-potassium hydrogen phosphate (K2HPO4) (Carl Roth, catalog number: 5066.1 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S9888 )
- Di-sodium hydrogen phosphate (Na2HPO4) (Carl Roth, catalog number: T876.1 )
- Bacto peptone (BD, catalog number: 211677 )
- Streptomycin sulfate salt (Sigma-Aldrich, catalog number: S6501 )
- Agar (Sigma-Aldrich, catalog number: 05040 )
- Cholesterol stock solution (SERVA Electrophoresis, catalog number: 17101.01 )
- Calcium chloride dihydrate (CaCl2·2H2O) (Sigma-Aldrich, catalog number: C5080 )
- Magnesium sulfate (MgSO4) (Sigma-Aldrich, catalog number: M7506 )
- Nystatin stock solution (Sigma-Aldrich, catalog number: N3503 )
- Agarose (Biozym, catalog number: 840004 )
- Phosphate buffer (1 M; sterile) (see Recipes)
- Nematode growth medium (NGM) agar plates (see Recipes)
- M9 buffer (see Recipes)
- 5% agarose pads (see Recipes)
Equipment
- Dissecting stereomicroscope (Olympus, model: SMZ645 )
- Epifluorescence microscope (ZEISS, model: Axio Imager Z2 , objective EC Plan-Neofluar 10x/0.3)
- Confocal microscope (we use the Zeiss LSM710 confocal microscope with an Argon multiline laser source 25 mW and a tunable laser with the wavelength range 488-640 nm) (ZEISS, model: LSM710)
- Microwave
- Incubators for stable temperature (AQUA®LYTIC incubator 20 °C)
- Scale
- Cylindrical glass beaker (25 ml) (VWR, catalog number: 213-1120 )
- Autoclave
Software
- ZEN 2009 software (or later), Carl Zeiss AG, Jena, Germany (or any other software controlling a fluorescence microscope or confocal microscope)
- Microsoft Office 2011 Excel (Microsoft Corporation, Redmond, USA)
- Fiji or ImageJ (https://fiji.sc/ or https://imagej.nih.gov/ij/)
Procedure
- Growth and synchronization of transgenic C. elegans population
- When working with a transgenic strain with integrated genetic array (Table 1), use a sterile pipette tip (200 µl) to cut a small chunk (0.5 x 0.5 cm) of agar containing animals from an older plate and transfer it to a freshly Escherichia coli (OP50) seeded NGM plate (see Recipes). When working with a transgenic line with a non-integrated extrachromosomal array, pick ~25 L4 larvae or adult transgenic animals based on the selection-marker to a freshly OP50 seeded NGM plate.
- Incubate the nematodes at the standard temperature of 20 °C.
- 3.5 days later the plates contain a mixed population of larval stages.
- Synchronize nematodes by picking ~30 transgenic animals in L4 stage under a dissecting stereomicroscope and transfer them onto separate OP50 seeded plates. The L4 stage can be identified based on the presence of a half-moon shaped light structure in the area of the vulva and the relative size of the animal (Figure 1).
- Grow the animals for 24 h at 20 °C into day 1 adults and proceed with mounting the sample (Procedure B).
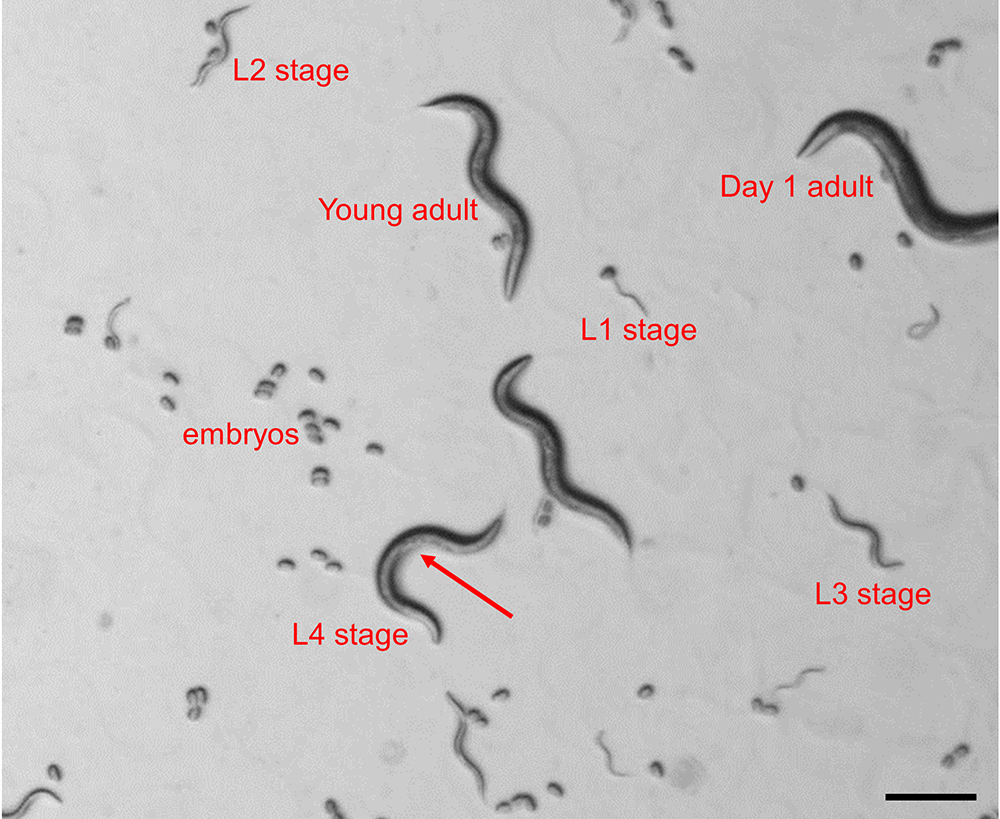
Figure 1. Identification of C. elegans larval stages in a mixed population. The image depicts all 4 larval stages, young adult and day 1 adult as seen under a dissecting stereomicroscope. The arrow indicates the half-moon shaped structure in the area of the vulva, which is indicative for the L4 larval stage.
- Mounting animals for imaging
- Prepare fresh 5% agarose pads (see Recipes).
- Pipette 3 µl polystyrene beads suspension in the center of the agarose pad.
- Use a platinum wire to pick ~30 transgenics from the OP50 seeded plates into the polystyrene bead suspension.
- Gently place a coverslip on top of the agarose/worm suspension and proceed with the imaging procedure. Optionally, the agarose pads can be sealed with nail polish, which will retain humidity for long-term (> 3 h) imaging.
- Animals can be recovered from unsealed plates by gently lifting one edge of the coverslip with the help of a scalpel and adding 10 µl of M9 buffer to suspend the worms. Subsequently, worms can be transferred with an eyelash to OP50-seeded NGM plates.
- Imaging P body/SG number with an epifluorescence light microscope
- Start up the light microscope and imaging software (ZEN, Zeiss).
- Place the agarose pad on the imaging stage of the fluorescent light microscope and locate the animals. Use a 10x objective to image the whole animal.
- Use the DIC channel to focus on an anatomical landmark. Most appropriate are the grinder and the lumen of the pharynx, which are located in the most central transverse (horizontal) position within the animal (Figure 2A).
- Change to the fluorescent channel to visualize P body or SG-specific fluorescence and define exposure time and fluorescent light intensity. These parameters depend on the transgene and have to be defined empirically. Saturation of the fluorescent signal has to be avoided by lowering exposure time and/or fluorescent light intensity. However, too short exposure might result in lowered sensitivity of detecting RNP granules (Figure 2B). For transgenes that co-express various reporters for P bodies/SGs repeat this step in the corresponding fluorescent channel.
- Once the best imaging conditions are determined, take a snapshot of all fluorescent channels and the DIC channel. Take images of at least 25 animals to obtain enough data for statistical analysis.
- Save the data as an image stack. Zeiss microscopes produce *.czi file formats, which can be processed by ImageJ/Fiji freeware (see Data analysis). However, images can also be stored/exported and further processed in any other common format including *.jpg, *.png, *.tiff, etc., and then proceed with the data analysis.
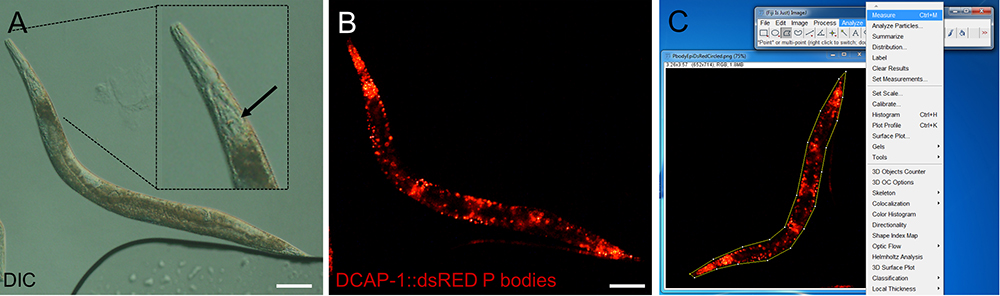
Figure 2. Representative images of epifluorescence images of an adult transgenic animal expressing the P body reporter DCAP-1::dsRED. Images were recorded with an Axio Imager Z2 through a 10x magnification objective. Size bars are 100 µm. A. DIC image. The inlay shows a digitally enhanced view on the pharynx. The arrow points out the grinder, which has to be in focus before switching to the fluorescent channel. B. Fluorescent image of the P body reporter DCAP-1::dsRED. C. Screenshot of image processing. Choose the polygon selection tool to surround the whole animal. Open Analyze > Measure to get area size and MPI. (Full genotype of transgenic animal: N2;Ex[pdcap-1DCAP-1::dsRED; pRF4] published in Rieckher et al., 2015.)
- Imaging P body/SG number, dynamics and colocalization with a confocal microscope
- Start up the confocal microscope and corresponding imaging software.
- Activate the appropriate laser(s) for fluorescent detection of P body/SG reporters.
- Locate the animal through the eyepiece with a 10x objective using transmission light and center the field of view on the area of interest (e.g., the pharynx).
- Upon localization, switch to the 40x objective and readjust the focus.
- In the confocal operating software (acquisition) design a protocol for a z-stack scan.
- Different RNP granules significantly vary in size and intensity across different transgenes and within samples (Teixeira et al., 2005). Hence, a balance has to be established empirically between laser power and gain (voltage of photomultiplier tubes [PMTs]/detectors) to set the highest and lowest detection limit. If available, use the Smart Setup as a starting point. Try to avoid overexposure but test the lower detection limit by stepwise increasing laser power and/or gain. The final settings have to be reproducibly applied to all samples. For the example in Figure 2 we use the tunable laser at 570 nm (excitation dsRED), laser power 3%, and Master gain 580.
- Keep the pinhole size for an optimal section thickness at 1 AU.
- The distance between z-stacks (slices) should be maximum 1.5 µm. Decreasing z-stack distance might enhance the capability to detect smaller RNP granules but increases photobleaching. Cover a similar volume in each scanned sample. We perform 30 z-stacks at 1.5 µm across the pharyngeal region (Figures 2 and 3).
- Assign the frame size (resolution). We use 1024 x 1024 (Figures 2 and 3).
- Use the transmitted light detector (T-PMT) to produce a DIC/Brightfield image in parallel (optional).
- Once the best imaging conditions are determined, perform a scan of the fluorescent channel. When imaging different P bodies/SGs co-expressed in the same transgene use sequential scan for additional fluorescent channels. Take datasets of at least 15 animals to obtain enough data for statistical analysis.
- Upon completion of the scan use the processing mode of the ZEN software to produce a maximum intensity projection (MIP), summarizing the fluorescence intensity of all stacks in one image (Figures 3A and 3B). Consistently save as *.czi file or other image file format. Proceed with data analysis. Alternatively, single stacks or subsets of stacks can be processed and analyzed.
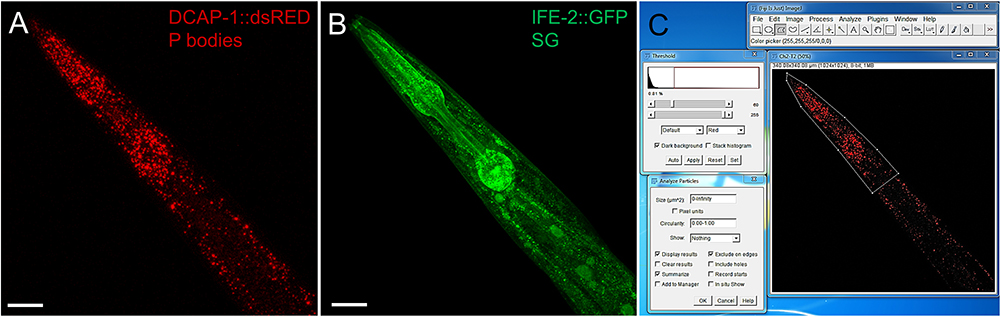
Figure 3 Representative images of the pharyngeal region of an adult transgenic animal expressing the P body reporter DCAP-1::dsRED and the SG reporter IFE-2::GFP. Images were recorded with an LSM710 confocal microscope and processed into MIP. Size bars are 50 µm. A. P body reporter DCAP-1::dsRED expression. B. SG reporter IFE-2::GFP. C. Screenshot of image processing via Fiji/ImageJ. Use the polygonal tool to surround the area of interest, assign a threshold and use Analyze Particles to obtain information about P body/SG intensity, number and size. (Full genotype of transgenic animal:N2;Ex[pdcap-1DCAP-1::dsRED; pife-2IFE-2::GFP; pRF4] published in Rieckher et al., 2015.)
Data analysis
- Processing P body- and SG-data with Fiji (ImageJ)
- Download and install the Fiji freeware from https://fiji.sc/.
- Open an image file from fluorescence microscopy or a MIP file from confocal laser scanning microscopy by dragging/dropping into the Fiji software.
- Images will open as stacks of DIC and the fluorescent channels that were recorded. In the control panel of Fiji chose Image > Stacks > Stack to images to split the channels.
- Change Image > Type > 8 bit for further analysis steps.
- Chose the image of the fluorescent channel that should be analyzed and apply the polygon selection tool to further narrow down the region of interest (ROI). For images received from fluorescence microscopy encircle the whole animal (Figure 2C). In MIP images a specific region, such as the pharynx, should be selected (Figure 3C).
- For epifluorescence images choose ‘Analyze > Measure’ to obtain a summary about the Area size and the mean pixel intensity (MPI) in arbitrary units (AU) of the assigned area. This information will be displayed in a separate window and can be copied/pasted directly from there in a data analysis software and/or saved as an excel file (*.xls).
- For confocal images proceed with the measurement of P body and SG number and size a threshold has to be determined that assigns single pixels or pixel clusters to define fluorescent RNP granules. Chose Image > Adjust > Threshold and empirically assign a proper Min/Max value (Figure 3C). This value will vary depending on the transgene, choice of microscope and the imaging settings. Once assigned, these values have to stay consistent throughout every analysis.
- Chose Analyze > Analyze Particles and tick ‘Display results’, ‘Summarize’ and ‘Exclude on Edges’ and press OK (Figure 3C).
- Two separate Windows display the ‘Results’ for every RNP granule, producing Area (Granule size) and Mean (MPI), and the ‘Summary’ including ‘Count’ (RNP granule number), ‘Total Area’ and ‘Average Size’. These data can be copied directly into a data analysis software and/or saved as excel file.
- Statistical analysis
- Stay consistent with the number of animals examined for each strain and condition.
- Each assay should be repeated at least three (3) times.
- Use the Mann Whitney or Wilcoxon Kruskal Wallis test with a significance cut-off level of P < 0.05 for comparisons between different groups and correct for multiple pairwise comparisons using Bonferroni or False Discovery Rate (FDR).
Notes
P bodies and SGs are known to substantially vary in size and number across cells (Teixeira et al., 2005). Measurements become more precise and final values underlie less standard deviation when focusing on quantification in specific tissues (e.g., muscles, pharynx, or intestine) and increasing sample size.
Recipes
- Phosphate buffer (1 M)
- For 1 L, dissolve 102.2 g KH2PO4 and 57.06 g K2HPO4 in distilled water and fill up to 1 L. This is a 1 M solution, pH 6.0
- Autoclave at 121 °C for 20 min
- Store at room temperature
- Nematode growth medium (NGM) agar plates
- Mix 3 g NaCl, 2.5 g Bacto peptone, 0.2 g streptomycin, 17 g agar and add 900 ml distilled water. Autoclave at 121 °C for 20 min
- Let cool to 55-60 °C
- Add 1 ml cholesterol stock solution, 1 ml 1 M CaCl2, 1 ml 1 M MgSO4, 1 ml nystatin stock solution, 25 ml sterile 1 M phosphate buffer, pH 6.0, and distilled sterile water up to 1 L
- Pipette 8 ml medium per Petri dish and leave to solidify
- Store the plates at 4 °C until use
- M9 buffer
- Dissolve 3 g KH2PO4, 6 g Na2HPO4, 5 g NaCl in 1 L distilled water
- Autoclave at 121 °C for 20 min
- Let cool and add 1 ml 1 M MgSO4 (sterile)
- Store M9 buffer at 4 °C
- 5% agarose pads
- Weigh 0.5 g agarose and add it into a cylindrical glass beaker
- Add 10 ml M9 buffer
- Heat the mixture in a microwave until it is close to boiling. Take it out, stir with a pipette tip and boil again. Repeat until the agarose is dissolved completely
- Modify two microscope slides by putting a stripe of tape along the midline
- Place an empty microscope slide between two taped slides (Figure 4A)
- Put a drop (ca. 50 µl) of fresh 5% agarose solution in the middle of the slide (Figure 4B)
- Take a fourth microscope slide and place it on top of the agarose drop. Gently press down to flatten the drop. The tape serves as spacer to give the agarose pad a specific thickness (Figure 4C)
- Let the agarose harden for 30 sec and remove the top microscope slide
- Quickly cut the edges of the agarose pad with the scalpel to a square of approximately 20 x 20 mm
- Immediately proceed with the sample preparation (see Procedure B), since the agarose pads will start drying within approximately 5 min
Note: Leaving the top microscope slide as a cover contains the humidity longer (approx. 1 h). Thus, several agarose pads can be prepared and used swiftly during the experiments.
Figure 4. Preparation of agarose pads for fluorescent imaging in C. elegans. A. Two glass slides modified with tape flank an empty microscope slide. B. Place a drop of 50 µl 5% agarose solution in the middle of the microscope slide (arrow). C. Swiftly put another microscope slide on top and gently push it down to flatten the agarose drop.
Acknowledgments
This work was funded by grants from the European Research Council (ERC), the European Commission 7th Framework Programme. We want to acknowledge the Fang-Yen laboratory, Philadelphia, PA, who developed the agarose pads for long-term imaging of C. elegans.
References
- Anderson, P. and Kedersha, N. (2008). Stress granules: the Tao of RNA triage. Trends Biochem Sci 33(3): 141-150.
- Buchan, J. R., Nissan, T. and Parker, R. (2010). Analyzing P-bodies and stress granules in Saccharomyces cerevisiae. Methods Enzymol 470: 619-640.
- Cornes, E., Porta-De-La-Riva, M., Aristizabal-Corrales, D., Brokate-Llanos, A. M., Garcia-Rodriguez, F. J., Ertl, I., Diaz, M., Fontrodona, L., Reis, K., Johnsen, R., Baillie, D., Munoz, M. J., Sarov, M., Dupuy, D. and Ceron, J. (2015). Cytoplasmic LSM-1 protein regulates stress responses through the insulin/IGF-1 signaling pathway in Caenorhabditis elegans. RNA 21(9): 1544-1553.
- Decker, C. J. and Parker, R. (2012). P-bodies and stress granules: possible roles in the control of translation and mRNA degradation. Cold Spring Harb Perspect Biol 4(9): a012286.
- Ding, L., Spencer, A., Morita, K. and Han, M. (2005). The developmental timing regulator AIN-1 interacts with miRISCs and may target the argonaute protein ALG-1 to cytoplasmic P bodies in C. elegans. Mol Cell 19(4): 437-447.
- Gallo, C. M., Munro, E., Rasoloson, D., Merritt, C. and Seydoux, G. (2008). Processing bodies and germ granules are distinct RNA granules that interact in C. elegans embryos. Dev Biol 323(1): 76-87.
- Huelgas-Morales, G., Silva-Garcia, C. G., Salinas, L. S., Greenstein, D. and Navarro, R. E. (2016). The stress granule RNA-binding protein TIAR-1 protects female germ cells from heat shock in Caenorhabditis elegans. G3 (Bethesda) 6(4): 1031-1047.
- Kedersha, N., Cho, M. R., Li, W., Yacono, P. W., Chen, S., Gilks, N., Golan, D. E. and Anderson, P. (2000). Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol 151(6): 1257-1268.
- Kedersha, N. L., Gupta, M., Li, W., Miller, I. and Anderson, P. (1999). RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2α to the assembly of mammalian stress granules. J Cell Biol 147(7): 1431-1442.
- Kim, E., Sun, L., Gabel, C. V. and Fang-Yen, C. (2013). Long-term imaging of Caenorhabditis elegans using nanoparticle-mediated immobilization. PLoS One 8(1): e53419.
- Rieckher, M., Kourtis, N., Pasparaki, A. and Tavernarakis, N. (2009). Transgenesis in Caenorhabditis elegans. Methods Mol Biol 561: 21-39.
- Rieckher, M., Kyparissidis-Kokkinidis, I., Zacharopoulos, A., Kourmoulakis, G., Tavernarakis, N., Ripoll, J. and Zacharakis, G. (2015). A customized light sheet microscope to measure spatio-temporal protein dynamics in small model organisms. PLoS One 10(5): e0127869.
- Rousakis, A., Vlanti, A., Borbolis, F., Roumelioti, F., Kapetanou, M. and Syntichaki, P. (2014). Diverse functions of mRNA metabolism factors in stress defense and aging of Caenorhabditis elegans. PLoS One 9(7): e103365.
- Sarov, M., Murray, J. I., Schanze, K., Pozniakovski, A., Niu, W., Angermann, K., Hasse, S., Rupprecht, M., Vinis, E., Tinney, M., Preston, E., Zinke, A., Enst, S., Teichgraber, T., Janette, J., Reis, K., Janosch, S., Schloissnig, S., Ejsmont, R. K., Slightam, C., Xu, X., Kim, S. K., Reinke, V., Stewart, A. F., Snyder, M., Waterston, R. H. and Hyman, A. A. (2012). A genome-scale resource for in vivo tag-based protein function exploration in C. elegans. Cell 150(4): 855-866.
- Sheth, U. and Parker, R. (2003). Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300(5620): 805-808.
- Sun, Y., Yang, P., Zhang, Y., Bao, X., Li, J., Hou, W., Yao, X., Han, J. and Zhang, H. (2011). A genome-wide RNAi screen identifies genes regulating the formation of P bodies in C. elegans and their functions in NMD and RNAi. Protein Cell 2(11): 918-939.
- Teixeira, D., Sheth, U., Valencia-Sanchez, M. A., Brengues, M. and Parker, R. (2005). Processing bodies require RNA for assembly and contain nontranslating mRNAs. RNA 11(4): 371-382.
- Updike, D. and Strome, S. (2010). P granule assembly and function in Caenorhabditis elegans germ cells. J Androl 31(1): 53-60.
- Zhang, L., Ding, L., Cheung, T. H., Dong, M. Q., Chen, J., Sewell, A. K., Liu, X., Yates, J. R., 3rd and Han, M. (2007). Systematic identification of C. elegans miRISC proteins, miRNAs, and mRNA targets by their interactions with GW182 proteins AIN-1 and AIN-2. Mol Cell 28(4): 598-613.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Rieckher, M. and Tavernarakis, N. (2017). P-body and Stress Granule Quantification in Caenorhabditis elegans. Bio-protocol 7(2): e2108. DOI: 10.21769/BioProtoc.2108.
- Rousakis, A., Vlanti, A., Borbolis, F., Roumelioti, F., Kapetanou, M. and Syntichaki, P. (2014). Diverse functions of mRNA metabolism factors in stress defense and aging of Caenorhabditis elegans. PLoS One 9(7): e103365.
Category
Developmental Biology > Cell signaling > Stress response
Cell Biology > Cell signaling > Stress response
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.