- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Transfer of Large Contiguous DNA Fragments onto a Low Copy Plasmid or into the Bacterial Chromosome
Published: Vol 6, Iss 22, Nov 20, 2016 DOI: 10.21769/BioProtoc.2002 Views: 11856
Reviewed by: Lionel SchiavolinKanika GeraAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Bacterial pathogenicity islands and other contiguous operons can be difficult to clone using conventional methods due to their large size. Here we describe a robust 3-step method to transfer large defined fragments of DNA from virulence plasmids or cosmids onto smaller autonomously replicating plasmids or directly into defined sites in the bacterial chromosome that incorporates endogenous yeast and λ Red homologous recombination systems. This methodology has been successfully used to isolate and integrate at least 31 kb of contiguous DNA and can be readily adapted for the recombineering of E. coli and its close relatives.
Background
The ability to isolate and propagate large pieces of DNA has vastly expanded the study of gene networks and operons. However, the traditionally used engineered plasmids for this purpose, such as bacterial artificial chromosomes (BACs), while extremely useful, are limited by problems with DNA stability, copy number, and complex assembly requirements. Alternatively, incorporating constructs directly into the bacterial chromosome provides advantages by both reducing variations in gene expression arising from the presence of multiple gene copies and ensuring stable maintenance of genes, while also avoiding the need for antibiotic selection.
The methodologies described here were originally designed to capture and transfer the 31 kb of DNA operons that encode the Shigella flexneri type 3 secretion system onto the Escherichia coli chromosome (Reeves et al., 2015). The procedure utilizes yeast homologous recombination to generate a capture vector, a plasmid that contains regions of DNA that flank the fragment to be transferred, followed by using the λ Red recombination system to transfer the region of DNA of interest from a large virulence plasmid or cosmid onto the capture vector. The introduction of unique ‘Landing Pad’ sequences flanking the target sequence can be used to transfer via site-specific recombination the region of DNA present on the capture vector to an experimentally defined location on the bacterial chromosome using a protocol previously established by Kuhlman and Cox (2010). The inclusion of flanking landing pad sequences does not preclude the propagation of the DNA of interest on an autonomously replicating plasmid, but rather affords the opportunity to subsequently introduce the captured DNA onto a defined site on the bacterial chromosome. While we favor the use of an engineered landing pad sequence, one could adapt the approach described below to target the insertion of the captured DNA to a specifically defined locus on the bacterial chromosome.
Materials and Reagents
- 1.7 ml microcentrifuge tubes
- Acid-washed, small glass beads, 425-600 μm (30-40 U.S. sieve) (Sigma-Aldrich, catalog number: G8772 )
- Large glass beads, 5 mm (Corning, catalog number: 7268-5 )
- Cell scrapers (Corning, Falcon®, catalog number: 353085 )
- Electroporation cuvettes (Thermo Fisher Scientific, Fisher Scientific, catalog number: FB101 )
- Petri dishes (100 x 15 mm) (VWR, catalog number: 89038-968 )
- ElectroMAXTM DH10β cells (Thermo Fisher Scientific, InvitrogenTM, catalog number: 18290015 )
- Saccharomyces cerevisiae yeast strain (BY4741) (GE Healthcare Dharmacon, catalog number: YS1048 or other ura3 minus strain)
- E. coli DH10β atp/gidB::Landing pad cassette. Tetracycline resistant strain harboring an integrated landing pad cassette (for use if transferring captured DNA into the chromosome) (Addgene, catalog number: 83036 )
- pLLX13 plasmid, or another suitable yeast/bacteria shuttle vector (Addgene, catalog number: 79825 )
- pLLX8 plasmid, or another suitable vector that encodes the desired antibiotic resistance cassette (Addgene, catalog number: 79838 )
- pKD46, temperature sensitive, λ red recombinase expression plasmid, or similar (Datsenko and Wanner, 2000)
- pTKred plasmid, temperature sensitive, bacterial expression vector for λ Red recombinase and I-SceI (Addgene, catalog number: 41062 )
- Gene specific primers (see Table 1)
- Yeast nitrogen base with ammonium sulfate (MP Biomedicals, catalog number: 114027-532 )
- CSM-URA, uracil dropout supplement (MP Biomedicals, catalog number: 4511-222 )
- D-glucose (Thermo Fisher Scientific, Fisher Scientific, catalog number: D16-10 )
- Agar (Thermo Fisher Scientific, Fisher Scientific, catalog number: BP1423-2 )
- L-(+)-arabinose (Sigma-Aldrich, catalog number: A3256-100 )
- Ampicillin sodium salt (Sigma-Aldrich, catalog number: A9518-25G )
- Kanamycin monosulfate (MP Biomedicals, catalog number: 02150029 )
- Tetracycline hydrochloride (Sigma-Aldrich, catalog number: T7660 )
- Spectinomycin dihydrochloride pentahydrate (Sigma-Aldrich, catalog number: S9007-5G )
- Distilled, deionized water
- Magnesium sulfate (MgSO4) (Sigma-Aldrich, catalog number: M2643-500G )
- KOD hot start DNA polymerase (EMD Millipore, catalog number: 71-086-3 )
- QIAquick Gel Extraction Kit (QIAGEN, catalog number: 28704 )
- NheI-HF® restriction enzyme (New England Biolabs, catalog number: R3131S )
- PmeI restriction enzyme (New England Biolabs, catalog number: R0560S )
- MluI-HF® restriction enzyme (New England Biolabs, catalog number: R3198S )
- Yeast extract (Thermo Fisher Scientific, Fisher Scientific, catalog number: BP1422-2 )
- Peptone (Thermo Fisher Scientific, Fisher Scientific, catalog number: BP1420-2 )
- Glycerol (Thermo Fisher Scientific, Fisher Scientific, catalog number: BP229-4 )
- Polyethylene glycol 3350 (PEG 3350) (Sigma-Aldrich, catalog number: 202444 )
- Lithium acetate (LiOAc) (Sigma-Aldrich, catalog number: 517992 )
- DNA sodium salt from salmon testes (Sigma-Aldrich, catalog number: D1626 ), resuspended in TE buffer (2 mg/ml)
- Tris-HCl, pH 8.0 (Promega, catalog number: H5123 )
- EDTA (Sigma-Aldrich, catalog number: E5134-500G )
- QIAprep Spin Miniprep Kit (QIAGEN, catalog number: 27104 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S3014-500G )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P5405-250G )
- Sodium acetate (NaOAc) (Sigma-Aldrich, catalog number: S2889 )
- Selective antibiotics
Kanamycin monosulfate (1,000x stock) (see Recipes)
Ampicillin sodium salt (1,000x stock) (see Recipes)
Tetracycline hydrochloride (1,000x stock) (see Recipes)
Spectinomycin dihydrochloride pentahydrate (1,000x stock) (see Recipes) - CSM-Ura selective media (see Recipes)
- YEPD media (see Recipes)
- TE buffer (see Recipes)
- SOB broth (see Recipes)
- SOC broth (see Recipes)
Equipment
- 100-150 ml Erlenmeyer flask
- Vortex (Scientific Industries, catalog number: SI-0136 )
- Incubators, set to 37 °C and 30 °C
- Water baths or heat blocks, set to 42 °C, 37 °C and 30 °C
- Microcentrifuge (Eppendorf, model: 5424 )
- Large centrifuge, able to handle up to 50 ml (for making competent yeast and bacteria)
- Electrophoresis system
- UV transilluminator
- Thermocycler (Bio-Rad Laboratories, model: PTC 200 ) or any other conventional thermocycler
- GenePulser II electroporation system (Bio-Rad Laboratories) or other electroporation device
- Nanodrop Lite spectrophotometer (Thermo Fisher Scientific, model: NanoDrop Lite ), or any other equipment/method suitable for quantifying DNA concentration
- Roller drum or shaker
Procedure
Part I: Generation of a capture vector
Note: The objective of this section is to create a capture vector (Figure 1), a plasmid that can subsequently be used to capture large defined fragments of DNA (Part II). At this point in designing a protocol, a decision needs to be made regarding whether or not to engineer the resulting capture vector to include flanking unique ‘landing pad’ sequences, a procedure needed later if the ultimate goal is to introduce the DNA fragment into a defined site within the bacterial chromosome using Landing Pad technology previously described in detail by Kuhlman and Cox (2010) (Part III).
- Preparation of DNA to be recombined to generate the capture vector
- Overview of DNA fragments
As outlined in Figure 1, the capture vector is assembled by combining the following 4 fragments of DNA via yeast endogenous homologous recombination: linearized vector, plus 3 PCR generated fragments of interest: targeting sequence 1 (TS1), targeting sequence 2 (TS2) and an ampicillin resistance (beta-lactamase) containing cassette.- Vector backbone: pLLX13 is a 9.94 kb yeast/E. coli shuttle vector that carries origins of replication and selection markers to support plasmid maintenance in bacteria (oriV-incP and tetRA) and yeast (CEN and URA3) as well as an origin of transfer (oriT) sequence which facilitates transfer between bacteria via conjugation (Wolfgang et al., 2003) (Figure 1C).
- Targeting sequences 1 (TS1) and 2 (TS2): 1 kb regions of DNA that share homology with the up- and downstream borders of the region intended to be captured and cloned. TS1 and TS2 are generated via PCR using primers that add 40 base pairs of homology to linearized pLLX13 on one end, and a spacer region containing several restriction sites on the other end (Figure 1A, Table 1). This can be accomplished via one round of PCR. However, if chromosomal integration of the captured region of DNA is desired, landing pad sequences that flank the targeting sequences should be incorporated into the PCR product. Nested PCR is used to insert these landing pad sequences between the targeting sequences and the pLLX13 homology regions (Figure 1B, Table 1).
Notes:- As represented in Figure 1C, the I-SceI restriction sites present in the capture vector that flank the landing pad sequences can be used as described in Kuhlman and Cox (2010) to liberate and target the capture DNA to a previously engineered landing pad site. See Part III and Figure 3.
- The spacer regions incorporated into the 3’ end of the TS1 PCR and the 5’ end of the TS2 PCR provide homologous DNA sequence for recombination with the ampicillin resistance cassette (below) and assembly of the capture vector. Additionally, the spacer regions contain consecutive restriction digest sites that are used to linearize the capture vector prior to recombination with target DNA sequences (Figure 2). The TS1 spacer region includes PmeI and HpaI restriction sequences and the TS2 spacer region includes MluI and PacI restriction sites (Figure 1C, Table 1). These restriction sites can be replaced with other restriction sites if desired by altering the primer sequence, i.e., if sites are contained in TS1 and/or TS2.
- As represented in Figure 1C, the I-SceI restriction sites present in the capture vector that flank the landing pad sequences can be used as described in Kuhlman and Cox (2010) to liberate and target the capture DNA to a previously engineered landing pad site. See Part III and Figure 3.
- AmpR cassette: region of DNA inserted between TS1 and TS2 to facilitate selection of the capture vector plasmid using bacterial transformation following assembly of the capture vector plasmid using yeast recombination. The AmpR carrying fragment from pLLX8 is amplified with primers that provide homology to the Target Sequences 1 and 2 spacer region sequences. The flanking homology on these DNA sequences enables their assembly by homologous recombination when co-transformed into competent S. cerevisiae.
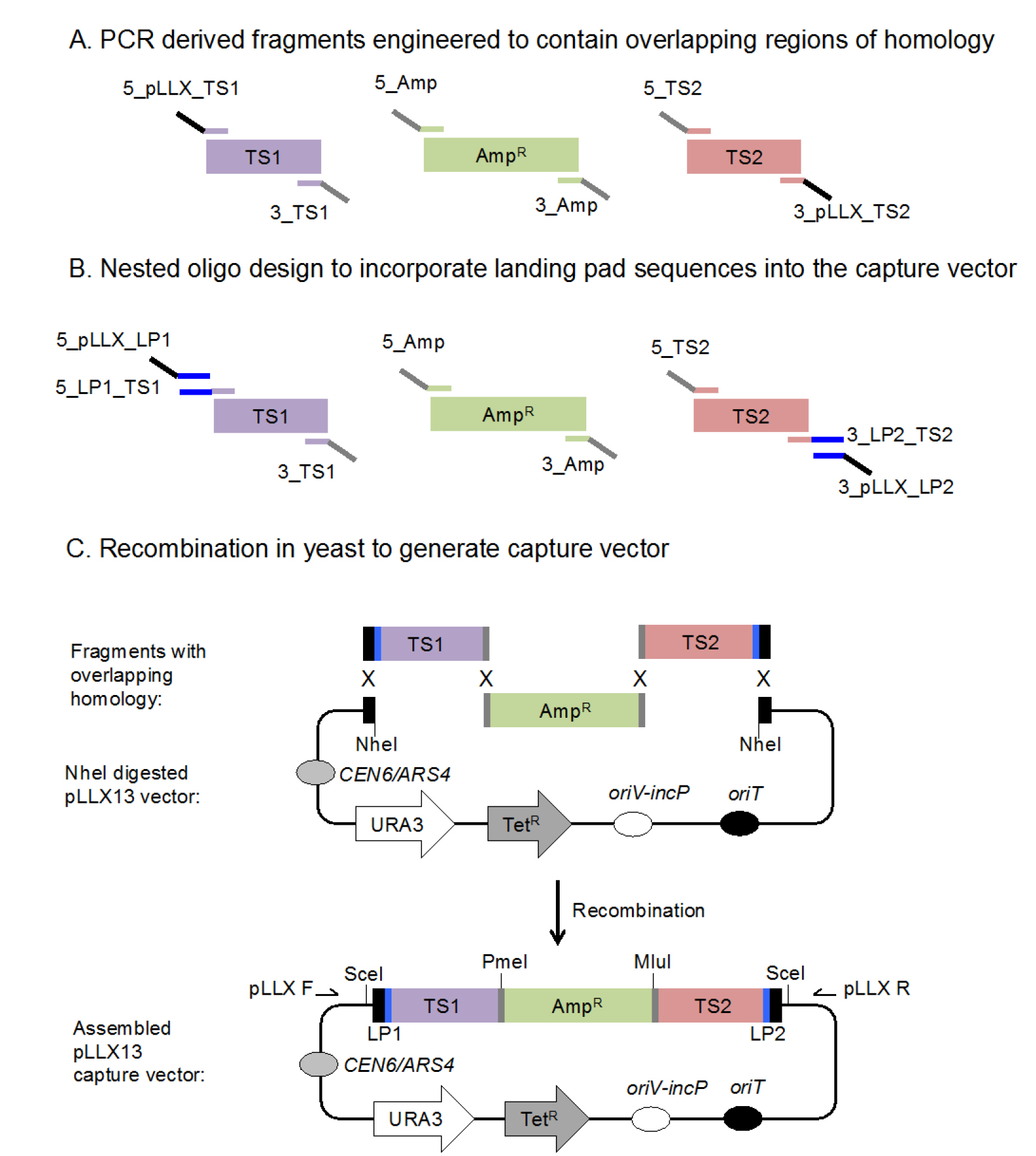
Figure 1. Schematic illustration of yeast recombination-based strategy utilized to generate a capture vector. A. Fragments with small overlapping regions of homology are generated via PCR using the depicted primers. The overlapping homologous sequences facilitate recombination and assembly in yeast. These fragments include those that contain homology to the 5’ and 3’ targeting sequences (TS1 and TS2) spacer regions (gray) plus an ampicillin resistance cassette (AmpR) (green). B. Landing pad (LP) sequences (blue) can be introduced into the capture vector by including a round of nested PCRs. The initial PCR amplification introduces homology to the spacer regions and flanking landing pad sequences, while a second round of PCR introduces homology to the pLLX13 vector backbone. C. When introduced into yeast, the 3 PCR generated fragments that contain overlapping homologous sequences and linearized (NheI-digested) pLLX13 vector backbone, undergo homologous recombination to generate a capture vector that can be selected for by growth on CSM-URA media. The resulting capture vector plasmid can be harvested and selected for in E. coli by plating on media containing ampicillin and tetracycline. The spacer regions (gray) that separate the target sequences and ampicillin resistance cassette introduce PmeI and MluI restriction sites into the capture vector which can be used to confirm proper capture vector assembly. Alternatively, SceI restriction sites can be used to liberate the entire assembled 5 kb fragment. Additionally, oligos pLLX F and pLLX R (Table 1), denoted by arrows, can be used to sequence the assembled capture vector to confirm that the proper recombination-mediated events occurred.
- Vector backbone: pLLX13 is a 9.94 kb yeast/E. coli shuttle vector that carries origins of replication and selection markers to support plasmid maintenance in bacteria (oriV-incP and tetRA) and yeast (CEN and URA3) as well as an origin of transfer (oriT) sequence which facilitates transfer between bacteria via conjugation (Wolfgang et al., 2003) (Figure 1C).
- Preparation of PCR generated fragments (TS1, TS2, AMPR)
- General PCR reaction mixture using high fidelity polymerase:
16 μl distilled water
2.5 μl KOD 10x reaction buffer
2.5 μl dNTPs (2 mM each)
1.5 μl 25 mM MgSO4
0.5 μl forward oligo 20 μM (see oligo descriptions below and in Table 1)
0.5 μl reverse oligo 20 μM
1 μl template DNA (50-250 ng for genomic DNA and 1-10 ng for plasmid DNA)
0.5 μl KOD hot start polymerase (1 U/μl) - Oligos and template DNA
Note: Choice of forward oligo (+/-LP) depends on whether downstream chromosomal integration of target DNA is desired.- TS1 PCR (+/-LP):
Forward oligo (+LP): 5_LP1_TS1 (Oligos are described in Table 1)
Forward oligo (-LP): 5_pLLX_TS1
Reverse oligo: 3_TS1
Template: Gene specific template such as bacterial chromosomal or virulence plasmid DNA - TS2 PCR (+/-LP):
Forward oligo: 5_TS2
Reverse oligo (+LP): 3_LP2_TS2
Reverse oligo (-LP): 3_pLLX_TS2
Template: Gene specific template such as bacterial chromosomal or virulence plasmid DNA - Ampicillin resistance cassette PCR:
Forward oligo: 5_Amp
Reverse oligo: 3_Amp
Template: pLLX8 or other plasmid containing desired antibiotic resistance cassette for selection - PCR profile:
Note: These conditions can be modified for the use of a different polymerase or for gene specific primers as needed.
1 cycle, 94 °C, 5 min
30 cycles, 94 °C 30 sec; 55 °C 30 sec; 72 °C 2-3 min
1 cycle, 72 °C 10 min
- TS1 PCR (+/-LP):
- Expected fragments
The TS1 and TS2 PCR products should be ~1 kb and the AmpR cassette ~3 kb. - Nested PCR (if adding flanking landing pad [LP] sequences)
Prepare the same KOD hot start polymerase PCR mixture as above with following primer and template modifications:- Nested TS1 PCR oligos:
Forward oligo: 5_pLLX_LP1
Reverse oligo: 3_TS1
Template: 1 μl of purified, confirmed TS1 PCR - Nested TS2 PCR oligos:
Forward oligo: 5_TS2
Reverse oligo: 3_pLLX_LP2
Template: 1 μl of purified, confirmed TS2 PCR
- Nested TS1 PCR oligos:
- Nested PCR analysis
The TS1 and TS2 nested PCR products are still approximately 1 kb, but now contain a region of homology to the pLLX13 vector backbone to be used in the downstream recombination reaction. - Clean the PCR products using the QIAquick Gel Extraction Kit or other equivalent methodology.
- General PCR reaction mixture using high fidelity polymerase:
- Preparation of linearized vector DNA
- Digest 1 μg of the pLLX13 vector with NheI-HF restriction enzyme.
- Gel purify the digested plasmid DNA and quantify concentration on a Nanodrop Lite spectrophotometer or other equipment/method suitable for DNA quantification.
Table 1. Primer sequences (5’-3’ sequence)
aThese sequences should be added to the 5’ end (TS1) or 3’ end (TS2) of gene specific sequence to yield PCR products that can easily recombine to form a capture vector in yeast.
bThese oligos contain PmeI and HpaI restriction sites as part of a spacer region.
cThese oligos contain PacI and MluI restriction sites as part of a spacer region.
NNN, denotes where target DNA specific sequence should be added.
Bold, Landing Pad sequence
Underlined, homology to pLLX13 vector
Italics, homology to pLLX8 ampicillin resistance cassette encoding vector. Sequences in italics can be substituted for vector of choice containing desired antibiotic resistance cassette.
- Digest 1 μg of the pLLX13 vector with NheI-HF restriction enzyme.
- Overview of DNA fragments
- Use of yeast recombination to generate capture vector
- Preparation of competent yeast (Gietz and Woods, 2002)
- Day 1: Inoculate 5 ml YEPD broth with a large colony of the host S. cerevisiae yeast strain and incubate on a roller drum overnight at 30 °C.
- Day 2: Measure the culture OD600 on a spectrophotometer, and back dilute (subculture) into 50 ml fresh YEPD broth in a 100-150 ml Erlenmeyer flask to an OD600 of 0.3.
- Incubate yeast on a shaker at 30 °C for 3-5 h until yeast cells undergo two doublings to reach an OD600 of ~1.2.
- Harvest yeast by centrifuging in a large centrifuge at 850 x g for 3 min. Decant supernatant.
Note: Centrifuging at a higher x g makes the yeast pellet more difficult to resuspend. - Resuspend yeast in 25 ml sterile water.
- Centrifuge at 850 x g for 3 min in a large centrifuge to pellet yeast. Decant supernatant.
- Resuspend yeast in 1 ml sterile water.
Note: Remaining yeast can be resuspended in 10% glycerol and frozen at -80 °C for future use.
- Day 1: Inoculate 5 ml YEPD broth with a large colony of the host S. cerevisiae yeast strain and incubate on a roller drum overnight at 30 °C.
- Yeast transformation
- Centrifuge 100 μl of competent yeast in microcentrifuge tube using tabletop centrifuge at 850 x g for 1 min and remove supernatant.
- Overlay the yeast with 240 μl filter sterilized 50% PEG 3350 (w/v).
Note: PEG 3350 is viscous, use plastic based materials to pipet. - Assemble transformation mixture composed of:
- 36 μl 1 M LiOAc
- 10 μl DNA sodium salt (ssDNA) from salmon testes (2 mg/ml), resuspended in TE buffer.
Note: Before first use, boil ssDNA for 5 min and cool on ice. - DNA transformation mixture:
100 ng linearized, NheI-digested, pLLX13 vector
200 ng targeting sequence 1 (TS1) amplified fragment
200 ng targeting sequence 2 (TS2) amplified fragment
600 ng amplified fragment that contains ampicillin resistance gene
Distilled, deionized water to a final volume of 65 μl
Note: A second DNA mixture can also be assembled as a control, which contains only 100 ng linearized, NheI-digested, pLLX13 vector. - Overlay the DNA transformation mixture upon the PEG and pipet or vortex until yeast pellet is fully resuspended.
- Incubate yeast at 30 °C for 30 min in a water bath or heat block.
- Transfer tube to a 42 °C water bath or heat block and incubate for an additional 15 min.
- Harvest cells by centrifuging at 2,400 x g for 1 min.
- Resuspend pellet in 100 μl sterile TE buffer or water and plate all onto a CSM-URA plate. Spread across plate using sterile large (5 mm) glass beads.
- Incubate in 30 °C incubator for up to 5 days. Colonies usually appear within 2-3 days, but growth times may vary.
- Isolate recombined capture vector
- Use a plate scraper or sterile pipet tip to resuspend all yeast colonies present on the transformation plate in 500 μl of YEPD media. Transfer mixture to a 1.7 ml microcentrifuge tube and harvest cells by centrifuging at 2,400 x g for 1 min.
- Decant supernatant and lyse yeast to extract assembled capture vectors using a QIAprep Spin MiniPrep Kit protocol modified as follows:
- Resuspend yeast in 250 μl of P1 buffer.
- Add 250 μl of small (425-600 μm) acid-washed glass beads to the tube and vortex on maximal speed setting for 5 min to lyse the yeast.
- Allow ~1 min for the glass beads to settle before transferring the yeast lysate into a new 1.7 ml microcentrifuge tube.
- Add 250 μl P2 buffer, mix by inversion 4-6 times and incubate at room temperature for 5 min.
- Add 350 μl N3 buffer, invert 4-6 times to mix, and spin at 16,500 x g for 10 min.
- Transfer supernatant onto a QIAprep column and collection tube and spin at 16,500 x g for 30 sec.
- Decant flow through and wash column with 750 μl PE wash buffer. Spin at 16,500 x g for 30 sec.
- Discard the flow through solution and reassemble spin column and collection tube. Spin at 16,500 x g for 1 min to dry the column.
- Transfer spin column to a fresh, labeled 1.7 ml microcentrifuge tube.
- Add 40 μl EB buffer (elution buffer), incubate at room temperature for 1 min, and perform a final spin at 16,500 x g for 1 min.
- Mix 3 μl of the eluted DNA with E. coli ElectroMAX DH10β cells (or any suitable competent E. coli cells) in a 1.7 ml microcentrifuge tube.
- Transfer DNA and bacteria mixture into a 0.1 mm electroporation cuvette.
- Electroporate using a GenePulser II (Bio-Rad Laboratories) or equivalent electroporation device using manufacturer’s settings.
Note: For the GenePulser II, use 1.8 kV and 25 μF settings with E. coli. - Add 450 μl of SOC and incubate for 1 h in a 37 °C heat block or water bath.
- Plate transformed bacteria on LB containing tetracycline and ampicillin to select for recombinant plasmids that have combined all 4 DNA fragments. Colonies will likely appear one day post-transformation, but can sometimes take an additional day to appear.
- To screen candidate capture vector colonies, inoculate at least 4 single colonies individually into 10 ml LB broth containing ampicillin and tetracycline and grow overnight at 37 °C.
Note: We recommend using a larger volume for the miniprep at this step (up to 10 ml), because the low copy of the pLLX13 vector backbone typically results in a lower DNA yield. The bacterial pellet from all 10 ml of culture can then be pooled into a single miniprep. - The next morning, harvest plasmid DNA using a QIAprep Spin Miniprep Kit following manufacturer’s instructions.
- Use a plate scraper or sterile pipet tip to resuspend all yeast colonies present on the transformation plate in 500 μl of YEPD media. Transfer mixture to a 1.7 ml microcentrifuge tube and harvest cells by centrifuging at 2,400 x g for 1 min.
- Preparation of competent yeast (Gietz and Woods, 2002)
- Data analysis Part I: Confirm correct assembly of capture vector
The plasmids isolated from individual bacterial transformants can be analyzed for the presence of an intact, recombined capture vector by:- Digesting with SceI to liberate the inserted (~5 kb) TS1-AmpR-TS2 fragment (Figure 1C).
- Digesting with PmeI and MluI to liberate the (~3 kb) AmpR cassette.
- Using PCR to confirm the presence of both TS1 and TS2.
- Sequencing the recombined junctions with oligos pLLX F and pLLX R (Table 1 and Figure 1).
- Digesting with SceI to liberate the inserted (~5 kb) TS1-AmpR-TS2 fragment (Figure 1C).
Part II: Capture the target sequence using λ Red homologous recombination
- Prepare capture vector DNA for transformation
Linearize the capture vector to generate a fragment of DNA such that TS1 and TS2 are accessible as free ends to promote recombination when introduced into bacteria. This will greatly improve the efficiency of λ Red recombination and acts to remove the ampicillin resistance cassette (AmpR) prior to recombination (Figure 2A).- Digest 1 μg pLLX13-TS1-amp-TS2 capture vector DNA with PmeI plus MluI.
Note: HpaI can be used as a substitute for PmeI, and PacI can be used as a substitute for MluI. - Gel purify the ~10 kb linearized capture vector DNA using a QIAquick Gel Extraction Kit purification kit according to manufacturer’s instructions.
- Quantify recovered linearized DNA using a Nanodrop Lite spectrophotometer or other DNA quantification method.
- Digest 1 μg pLLX13-TS1-amp-TS2 capture vector DNA with PmeI plus MluI.
- Introduce source of target DNA to be captured into E. coli ElectroMAX DH10β cells
For the following protocol, the target DNA must first be engineered to contain a selectable marker to facilitate selection of recombination based capture events. For example, the Shigella virulence plasmid contained a KanR cassette in place of ipaJ (Reeves et al., 2015).
Notes:- If removal of the antibiotic resistance cassette is desired for downstream applications, FRT (FLP recognition target) sites can be included that flank the antibiotic resistance cassette as described in Datsenko and Wanner (2000). It may be possible to capture an unmarked region of DNA via recombination, however this possibility was not investigated here.
- It is not absolutely necessary to transfer the DNA source into E. coli, if the DNA source is present in a bacterial species that can efficiently perform λ Red recombination. However, in our experience, performing this step greatly improved the efficiency and success of DNA capture.
- Transform marked cosmid or virulence plasmid into ElectroMAX DH10β cells via electroporation and select on LB plates containing kanamycin (or suitable antibiotic resistance marker).
Note: Only with these commercially available cells have we successfully introduced the large 220 kb Shigella virulence plasmid into a laboratory strain of E. coli.
- If removal of the antibiotic resistance cassette is desired for downstream applications, FRT (FLP recognition target) sites can be included that flank the antibiotic resistance cassette as described in Datsenko and Wanner (2000). It may be possible to capture an unmarked region of DNA via recombination, however this possibility was not investigated here.
- Use λ Red recombination system to capture DNA sequence of interest (Figure 2B).
- To enable the expression of a homologous recombination system in the target bacterial strain, the plasmid pKD46 (Datsenko and Wanner, 2000) is first introduced into the strain by standard electroporation and selection on solid media containing ampicillin and 0.2% glucose (to suppress λ Red expression) at 30 °C.
Note: pKD46 is a temperature sensitive, ampicillin resistant plasmid that carries an allele of λ red recombinase under the control of an arabinose-inducible promoter. Other plasmids encoding λ Red recombinase can also be used (Murphy and Campellone, 2003).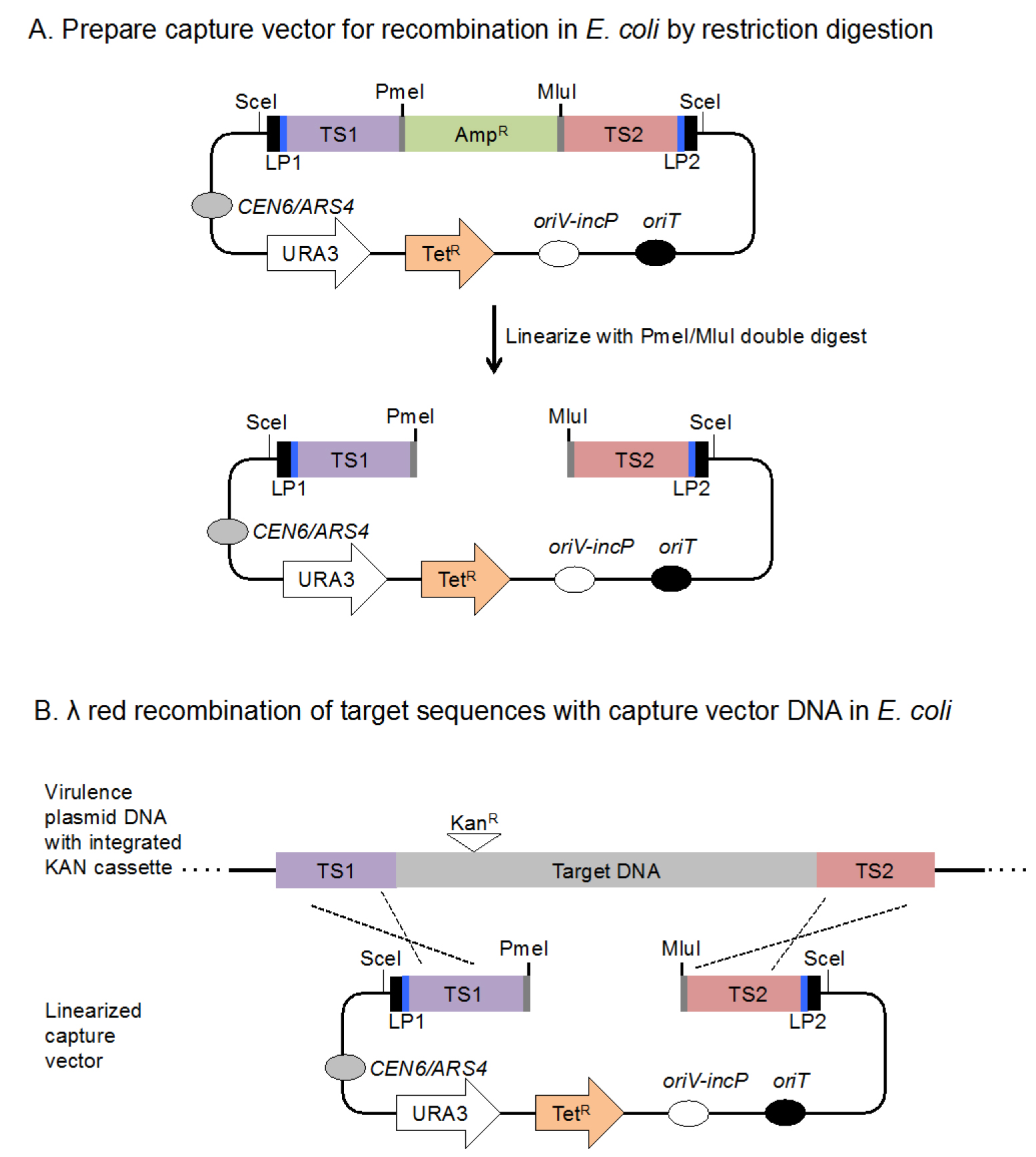
Figure 2. Schematic illustrating how to use the capture vector to isolate a target DNA sequence onto the yeast-generated capture vector. A. The capture vector is linearized by digestion with PmeI and MluI to generate free DNA ends corresponding to TS1 and TS2 DNA to enhance recombination efficiency and to remove the ampicillin resistance cassette. B. The linearized DNA is introduced into an E. coli strain that contains a virulence plasmid or cosmid which encodes the target DNA sequence engineered to contain a selectable marker, i.e., a kanamycin resistance cassette, plus a plasmid that conditionally expresses the λ Red recombinase,i.e., pKD46 (not shown). The previous introduction of an antibiotic selection marker into the target DNA facilitates the selection of the DNA fragment following its integration into the capture vector. Expression of λ Red recombinase results in the transfer of the target DNA onto the linearized capture vector via homologous recombination (dashed lines). - Day 1: Inoculate a single kanamycin, ampicillin resistant colony of bacteria that carries both the target DNA and pKD46 into LB broth containing kanamycin, ampicillin, and 0.2% glucose and grow overnight on a roller drum at 30 °C.
- Day 2: In the morning, back dilute the pKD46 containing bacteria 1:100 into 50 ml of SOB broth that contains kanamycin, ampicillin, and 0.2% arabinose (to induce λ Red recombinase expression). Incubate at 30 °C on a shaker for approximately 3-4 h until the culture reaches an OD600 of 0.6.
- Generate electrocompetent cells
- Transfer the cells into a sterile bottle and centrifuge at 6,000 x g for 10 min at 4 °C.
- Decant the supernatant and resuspend bacteria in 25 ml ice cold 10% glycerol.
Note: Keep the bacterial pellets cold through entire procedure by placing tubes on ice in between wash steps. - Repeat centrifugation at 6,000 x g for 10 min.
- Decant supernatant and repeat the 10% glycerol wash 3 more times, for a total of 4 washes.
Note: The 4 washes may be performed with ice-cold sterile water instead of 10% glycerol, if desired (steps C6-C8). - Resuspend the final pellet in 500 μl of cold 10% glycerol.
- Transfer 100 μl of electrocompetent, recombinase-expressing cells into a new microcentrifuge tube.
- Add 100 ng of linearized, gel purified, capture vector (Figure 2) and transfer mixture of bacterial cells and DNA into a 0.1 mm cuvette.
- Electroporate using a GenePulser II (Bio-Rad Laboratories) or equivalent electroporation device following manufacturer’s settings.
- Add 450 μl SOC broth to the electroporated cells and transfer to a microcentrifuge tube.
- Incubate cells at 37 °C for 1 h on a heat block or in a water bath.
- Plate entire transformation on LB plates containing tetracycline (to select for the capture vector backbone), and kanamycin or an appropriate antibiotic (to select for the region of target DNA to be captured) and incubate at 37 °C overnight.
Notes: - If no colonies arise after overnight incubation, continue incubating for an additional 24 h, as sometimes the recombination process can slow growth of the colonies.
- Incubation at 37 °C should eliminate the temperature sensitive pKD46 λ Red recombinase expressing plasmid, resulting in transformants that are ampicillin susceptible. The loss of pKD46 can be confirmed by patching individual colonies on LB containing ampicillin, if desired.
- Inoculate several candidate tetracycline/kanamycin resistant colonies into 10 ml of LB broth containing kanamycin and tetracycline and grow overnight at 37 °C.
- The next day, harvest DNA using a QIAprep Spin Miniprep Kit or other suitable miniprep kits.
Note: This step provides size exclusion of the recombined ‘captured’ DNA-containing plasmids away from the virulence plasmid and genomic DNA. - Transform harvested plasmids into ElectroMAX DH10β cells or other suitable electrocompetent bacterial host strain and plate on LB plates containing tetracycline and kanamycin.
Notes:- We recommend ElectroMAX DH10β cells for this purpose, as these cells are efficient at transformation and propagation of large DNA constructs.
- Importantly, if pursuing integration of the captured target DNA into the landing pad site of a bacterial chromosome, the harvested plasmids can be directly transformed into a landing pad-containing host strain (see Part III). However, if the user is unable to successfully transform the landing pad host strain directly, (i.e., because the plasmid containing captured target DNA is very large), we recommend transforming ElectroMAX DH10β cells first, and then using conjugation to transfer the plasmid into the landing pad-containing host strain.
- Once transformed into a host strain, the confirmed large plasmid can be transferred between bacterial strains via DNA conjugation, if desired.
- We recommend ElectroMAX DH10β cells for this purpose, as these cells are efficient at transformation and propagation of large DNA constructs.
- To enable the expression of a homologous recombination system in the target bacterial strain, the plasmid pKD46 (Datsenko and Wanner, 2000) is first introduced into the strain by standard electroporation and selection on solid media containing ampicillin and 0.2% glucose (to suppress λ Red expression) at 30 °C.
- Data analysis Part II: Confirm transfer of the target DNA segment onto the capture vector
- We recommend using several complementary approaches to confirm that the correct target DNA segment has been transferred onto the capture vector. In our experience, nearly 100% of plasmids recovered from E. coli that carry both antibiotic markers contain the correct target DNA fragment.
- Candidate plasmids should be screened for the correct recombination event by:
- PCR analysis of junction regions (i.e., PCR using the pLLXF or pLLXR oligo [Table 1 and Figure 1]) and a suitable target DNA oligo.
- PCR analysis using gene specific primers for sequences that should be included in the captured region of DNA. Positive and negative controls should be included, for example, using the empty capture vector as PCR template for the negative control, and the parent bacterial strain that contains the target DNA segment as a positive control.
- PCR reactions using primers that specifically bind to regions unique to the starting virulence plasmid or cosmid, but not present in the captured DNA region, can be performed to confirm loss of the parental virulence plasmid or cosmid.
- Sequence analysis of the captured target DNA using target gene specific oligos, pLLXF, or pLLXR.
- PCR analysis of junction regions (i.e., PCR using the pLLXF or pLLXR oligo [Table 1 and Figure 1]) and a suitable target DNA oligo.
- We recommend using several complementary approaches to confirm that the correct target DNA segment has been transferred onto the capture vector. In our experience, nearly 100% of plasmids recovered from E. coli that carry both antibiotic markers contain the correct target DNA fragment.
Part III: Integration of target DNA into a landing pad site on the bacterial chromosome
Notes:
- This section describes how to transfer a large piece of target DNA from the capture vector, assembled in Part II, into the chromosome of a bacterial strain that harbors a landing pad site. The introduction of a landing pad site into the bacterial chromosome is described in detail in the methods paper by Kuhlman and Cox (2010), and is therefore not discussed here. Thus, to continue with Part III of this protocol, a bacterial strain, such as E. coli DH10β, that harbors an integrated landing pad site is required.
- E. coli DH10β, that harbors an integrated landing pad site at the atp/gidB locus, is available at Addgene, catalog number: 83036. The landing pad site also introduces a tetracycline resistance cassette into the chromosome (Figure 3).
- Preparation of the bacterial strain for integration of captured, target DNA into the chromosome at a landing pad site.
Notes:- In order to transfer target DNA into the landing pad site of a bacterial chromosome, the captured target DNA plasmid must contain flanking landing pad sequences introduced by nested PCR (see Part I and Figure 1B).
- The landing pad host strain must contain two plasmids:(1) the pLLX13 capture vector containing the captured target DNA sequences (generated and confirmed in Part II) and (2) pTKred, a spectinomycin resistant, temperature sensitive plasmid that expresses an IPTG-driven λ red recombinase and an arabinose driven SceI restriction endonuclease. This protocol uses E. coli DH10β harboring a landing pad site at the atpI/gidB locus. If the captured target DNA plasmid (from Part II) was already introduced into E. coli DH10β-landing pad in Part II, begin this protocol by introducing pTKred into the strain by going directly to step B (Introduce the pTKred plasmid into E. coli DH10β-landing pad bacteria containing the captured target DNA).
- Introduce the capture vector plasmid containing target DNA into E. coli DH10β-landing pad cells
- Day 1: Inoculate a single tetracycline resistant E. coli DH10β-landing pad colony into LB broth containing tetracycline and grow overnight on a roller drum at 37 °C.
- Day 2: In the morning, back-dilute the E. coli DH10β-landing pad bacteria 1:100 into 10 ml of LB broth that contains tetracycline. Incubate at 37 °C on a roller drum or shaker for approximately 1-2 h until the culture reaches an OD600 of 0.6.
- Generate electrocompetent E. coli DH10β-landing pad cells:
- Incubate the cells on ice for 1 h.
- Transfer the cells into a sterile bottle and centrifuge at 6,000 x g for 10 min at 4 °C.
- Decant the supernatant and resuspend bacteria in 5 ml ice cold 10% glycerol.
Note: Keep the bacterial pellets cold through entire procedure by placing tubes on ice in between wash steps. - Repeat centrifugation at 6,000 x g for 10 min.
- Decant supernatant and repeat the 10% glycerol wash 3 more times, for a total of 4 washes.
Note: The 4 washes may be performed with ice-cold sterile water instead of 10% glycerol, if desired. - Resuspend the final pellet in 200 μl of cold 10% glycerol.
- Incubate the cells on ice for 1 h.
- Transfer 100 μl of electrocompetent cells into a new microcentrifuge tube.
- Add 200 ng of plasmid containing captured target DNA (Figure 2B) and transfer mixture of bacterial cells and DNA into a 0.1 mm cuvette.
- Electroporate using a GenePulser II (Bio-Rad Laboratories) or equivalent electroporation device following manufacturer’s settings.
- Add 450 μl SOC broth to the electroporated cells and transfer to a microcentrifuge tube.
- Incubate cells at 37 °C for 1 h on a heat block or in a water bath.
- Plate entire transformation on LB plates containing tetracycline (to select for the landing pad site on the chromosome), and kanamycin or an appropriate antibiotic (to select for the region of captured target DNA) and incubate at 37 °C overnight.
- Day 1: Inoculate a single tetracycline resistant E. coli DH10β-landing pad colony into LB broth containing tetracycline and grow overnight on a roller drum at 37 °C.
- Introduce the pTKred plasmid into E. coli DH10β-landing pad bacteria containing the captured target DNA
- Day 1: Inoculate a single colony of tetracycline/kanamycin resistant E. coli DH10β-landing pad bacteria containing the captured target DNA plasmid into LB broth containing tetracycline and kanamycin and grow overnight on a roller drum at 37 °C.
- Day 2: In the morning, back dilute the E. coli DH10β-landing pad bacteria containing the captured target DNA plasmid 1:100 into 10 ml of LB broth that contains tetracycline and kanamycin. Incubate at 37 °C on a roller drum or shaker for approximately 1-2 h until the culture reaches an OD600 of 0.6.
- Generate electrocompetent cells
- Incubate the cells on ice for 1 h.
- Transfer the cells into a sterile bottle and centrifuge at 6,000 x g for 10 min at 4 °C.
- Decant the supernatant and resuspend bacteria in 5 ml ice cold 10% glycerol.
Note: Keep the bacterial pellets cold through entire procedure by placing tubes on ice in between wash steps. - Repeat centrifugation at 6,000 x g for 10 min at 4 °C.
- Decant supernatant and repeat the 10% glycerol wash 3 more times, for a total of 4 washes.
Note: The 4 washes may be performed with ice-cold sterile water instead of 10% glycerol, if desired. - Add 100 ng of pTKred plasmid and transfer mixture of bacterial cells and DNA into a 0.1 mm cuvette.
- Electroporate using a GenePulser II (Bio-Rad Laboratories) of equivalent electroporation device following manufacturer’s settings.
- Add 450 μl SOC broth to the electroporated cells and transfer to a microcentrifuge tube.
- Incubate cells at 30 °C for 1 h.
Note: pTKred is temperature sensitive so the outgrowth and plating must be performed at 30 °C. - Plate entire transformation on LB plates containing tetracycline (to select for landing pad on the chromosome), kanamycin (to select for the captured target DNA plasmid), spectinomycin (to select for pTKred), and 0.2% D-glucose (to inhibit expression of λ Red recombinase) and incubate overnight at 30 °C.
- Day 1: Inoculate a single colony of tetracycline/kanamycin resistant E. coli DH10β-landing pad bacteria containing the captured target DNA plasmid into LB broth containing tetracycline and kanamycin and grow overnight on a roller drum at 37 °C.
- Insertion of target DNA into the landing pad on the chromosome
Notes:- This part of the protocol is modified from Kuhlman and Cox (2010). During this step, arabinose is used to induce expression of the SceI restriction enzyme while IPTG induces expression of λ Red recombinase. SceI introduces double stranded DNA breaks into the E. coli DH10β chromosome at the site of the landing pad integration. Simultaneously, SceI introduces double stranded DNA breaks on the captured target DNA plasmid. Expression of λ Red recombinase leads to the homologous recombination between landing sites on the chromosome and those present at the free ends of the target DNA. Successful SceI digestion and homologous recombination lead to double stranded break repair of the chromosome, excision of the tetracycline cassette, and integration of the target DNA sequence in its place (Figure 3). The introduction of double stranded breaks generates free ends of DNA that greatly increases the efficiency of homologous recombination.
- The 18 bp SceI restriction recognition site does not naturally exist in the E. coli chromosome.
- Day 1: In the morning, inoculate 5 ml of SOB broth containing 0.5% glycerol, 2 mM IPTG, and 0.2% arabinose with a single colony of the E. coli DH10β-landing pad strain containing the captured target DNA plasmid and pTKred. Grow for 1 h at 37 °C on a roller drum or shaker.
- Add 5 μl of spectinomycin (100 mg/ml) to the broth and incubate on a roller drum or shaker at 30 °C for 4 h.
- Add 5 μl of kanamycin (100 mg/ml) to the broth and incubate on a roller drum or shaker at 30 °C overnight.
- Day 2: Make serial dilutions of the overnight culture in LB broth from 10-1 to 10-5 and plate 100 μl of cells onto LB plates containing kanamycin.
- Incubate plates overnight at 37 °C.
- Day 3: Patch kanamycin resistant colonies onto LB plates containing tetracycline and LB plates containing kanamycin to screen for colonies that have become tetracycline susceptible.
- Incubate plates overnight at 37 °C.
- Colonies that are kanamycin resistant and tetracycline susceptible are candidates for successful target DNA integration at the landing pad site. (i.e., the tetracycline resistance cassette present on the chromosome has been replaced by the target DNA sequence).
Note: Incubation at 37 °C should also eliminate the temperature sensitive pTKred plasmid. Loss of pTKred can be confirmed by patching colonies onto LB plates containing spectinomycin. If growth at 37 °C is not sufficient to drop the pTKred plasmid, individual colonies can be grown for 4 h or overnight at 42 °C and then replated onto LB plates containing kanamycin at 37 °C, then retested for spectinomycin sensitivity. Passaging at the higher temperature should ensure loss of pTKred.
- This part of the protocol is modified from Kuhlman and Cox (2010). During this step, arabinose is used to induce expression of the SceI restriction enzyme while IPTG induces expression of λ Red recombinase. SceI introduces double stranded DNA breaks into the E. coli DH10β chromosome at the site of the landing pad integration. Simultaneously, SceI introduces double stranded DNA breaks on the captured target DNA plasmid. Expression of λ Red recombinase leads to the homologous recombination between landing sites on the chromosome and those present at the free ends of the target DNA. Successful SceI digestion and homologous recombination lead to double stranded break repair of the chromosome, excision of the tetracycline cassette, and integration of the target DNA sequence in its place (Figure 3). The introduction of double stranded breaks generates free ends of DNA that greatly increases the efficiency of homologous recombination.
- Data analysis Part III: Confirm integration of target DNA into the chromosomal landing pad site
Individual colonies can be analyzed for the presence of integrated target DNA by:- PCR analysis using primers to assess integration at junction regions in the chromosome. If the E. coli DH10β-landing pad strain was used, the atpI/gidB F and atpI/gidB R oligos (Figure 3 and Table 1) can be used with target gene specific primers to assess junctions.
- PCR analysis using primers to assess presence or absence of expected target DNA genes in the integrant strains.
- Functional analysis of integrated target DNA genes can be performed, if appropriate.
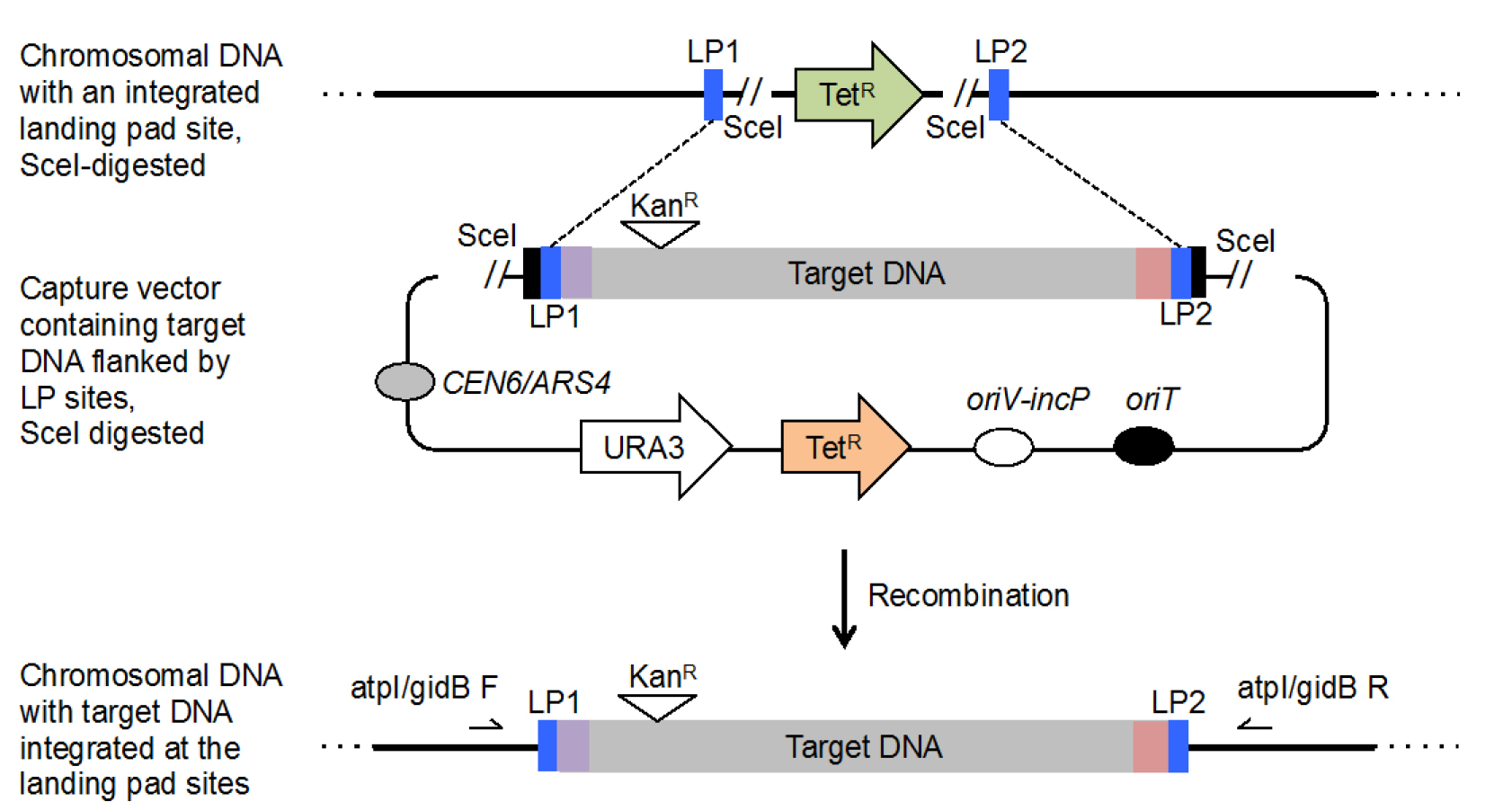
Figure 3. Schematic illustrating the transfer of target DNA from the capture vector into a bacterial chromosome at a landing pad site. Captured, target DNA sequences can be integrated into the chromosome of a bacterial host strain that harbors an integrated landing pad site. Landing pad site 1 (LP1) and 2 (LP2) are shown as blue boxes. Two plasmids must be introduced into the host strain: (1) the capture vector containing target DNA flanked by landing pad sites and SceI restriction sites (see Part I and Table 1 for design) and (2) pTKred, which encodes IPTG inducible λ Red recombinase and arabinose inducible SceI restriction endonuclease (not shown). Induction of SceI with arabinose introduces double stranded DNA breaks into the host strain chromosome and on the captured target DNA adjacent to the landing pad sites. The free ends of DNA are then joined by homologous recombination through the IPTG-induced expression of λ Red recombinase (dashed lines). Bacterial isolates containing the correct insertion of target DNA can be identified by screening for kanamycin resistance and tetracycline susceptibility, which indicates a loss of both the landing pad intervening sequence from the chromosome and the original capture vector containing target DNA. The original 1 kb targeting sequence 1 (TS1) is denoted by a purple box and targeting sequence 2 (TS2) is denoted by a red box. The original pLLX13 homology sequences are denoted as black boxes. PCR analysis with the atpI/gidB F or atpI/gidB R oligos, denoted by arrows, (Table 1) and target DNA specific oligos can be used to confirm integration at the landing pad site.
- PCR analysis using primers to assess integration at junction regions in the chromosome. If the E. coli DH10β-landing pad strain was used, the atpI/gidB F and atpI/gidB R oligos (Figure 3 and Table 1) can be used with target gene specific primers to assess junctions.
Notes
- Additional application: Capturing fragments of chromosomal DNA can be accomplished by modifying this protocol as described in Wolfgang et al. (2003). For example, a capture vector can be designed to include targeting sequences homologous to regions on the bacterial chromosome.
- Repetitive elements in DNA. An effort should be made when designing the initial capture vector targeting sequences (TS1 and TS2) to ensure the 1 kb of target sequence is unique and does not include a repetitive element or transposon sequence, as this can affect the specificity of the λ Red recombination reaction.
Recipes
- Kanamycin monosulfate (1,000x stock)
30 mg/ml in dH2O, filter sterilize - Ampicillin sodium salt (1,000x stock)
100 mg/ml in dH2O, filter sterilize - Tetracycline hydrochloride (1,000x stock)
12.5 mg/ml in 100% methanol, filter sterilize - Spectinomycin dihydrochloride pentahydrate (1,000x stock)
100 mg/ml in dH2O, filter sterilize - CSM-URA selective media (per liter 900 ddH2O)
6.7 g yeast nitrogen base with ammonium sulfate
0.77 g CSM-URA, uracil dropout supplement
~20 g agar
Before pouring plates add 100 ml sterile 20% glucose - YEPD (per liter ddH2O)
10 g yeast extract
20 g peptone)
20 g D-glucose
~15 g agar - TE buffer
10 mM Tris-HCl, pH 8.0
1.0 mM EDTA - SOB broth (per liter ddH2O)
5 g yeast extract
20 g tryptone
0.584 g NaCl
0.186 g KCl
2.4 g MgSO4
Adjust to pH 7.5 before use - SOC broth (per liter ddH2O)
Add 10 ml sterile 20% D-glucose solution to 990 ml SOB broth
Acknowledgments
This protocol was adapted from Reeves et al. (2015). Work was supported by R01AI064285, R21AI103882, and the Massachusetts General Hospital Research Scholar Award 2016 to CFL. AZR is supported by an MGH ECOR Fund for Medical Discovery Fellowship.
References
- Datsenko, K. A. and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97(12): 6640-6645.
- Gietz, R. D. and Woods, R. A. (2002). Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol 350: 87-96.
- Kuhlman, T. E. and Cox, E. C. (2010). Site-specific chromosomal integration of large synthetic constructs. Nucleic Acids Res 38(6): e92.
- Murphy, K. C. and Campellone, K. G. (2003). Lambda red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol Biol 4: 11.
- Reeves, A. Z., Spears, W. E., Du, J., Tan, K. Y., Wagers, A. J. and Lesser, C. F. (2015). Engineering Escherichia coli into a protein delivery system for mammalian cells. ACS Synth Biol 4(5): 644-654.
- Wolfgang, M. C., Kulasekara, B. R., Liang, X., Boyd, D., Wu, K., Yang, Q., Miyada, C. G. and Lory, S. (2003). Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 100(14): 8484-8489.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Reeves, A. Z. and Lesser, C. F. (2016). Transfer of Large Contiguous DNA Fragments onto a Low Copy Plasmid or into the Bacterial Chromosome. Bio-protocol 6(22): e2002. DOI: 10.21769/BioProtoc.2002.
Category
Microbiology > Microbial genetics > Gene mapping and cloning
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.