- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Determination of (p)ppGpp Levels During Stringent Response in Streptomyces coelicolor by Thin Layer Chromatography


Published: Vol 6, Iss 21, Nov 5, 2016 DOI: 10.21769/BioProtoc.1995 Views: 11233
Reviewed by: Valentine V TrotterDaan C. SwartsModesto Redrejo-Rodriguez
Original research article
The authors used this protocol in:

May 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
The stringent response in bacteria is a stress response that is mediated by the signaling molecules guanosine tetraphosphate and pentaphosphate [(p)ppGpp], alarmones that are also directly related to virulence. Therefore, determination of (p)ppGpp levels is crucial for studying the stringent response. The protocol here outlines in a step-wise manner the detection of (p)ppGpp in the bacterium Streptomyces coelicolor during stringent response (Strauch et al., 1991) by thin layer chromatography (TLC). In the example shown here, stringent response is induced by addition of serine hydroxamate, an inhibitor of seryl tRNA synthetase. This protocol was first published in Molecular Microbiology (Sivapragasam and Grove, 2016).
Background
Thin layer chromatography has been used for analyzing (p)ppGpp levels during stringent response in various bacterial species for a long time, and it is a generally accepted method for this purpose. However, previously published protocols only summarized the main concepts, and it was challenging to identify a comprehensive protocol that comprised every step of the procedure. We present here a detailed protocol that has been optimized for studying stringent response in S. coelicolor. Steps unique to handling of S. coelicolor cultures have been identified, and the protocol can therefore be readily adapted to other bacterial species. The method relies on the use of TLC plates that incorporate polyethyleneimine (PEI), which is a strong basic anion exchanger. PEI is therefore the matrix of choice for separation of ionic compounds such as phosphorylated nucleosides (Calderón-Flores et al., 2005; Mechold et al., 2013; Strauch et al., 1991).
Materials and Reagents
- Polyethyleneimine (PEI)-cellulose TLC plates (Sigma-Aldrich, catalog number: Z122882 )
- 1.5 ml sterile microcentrifuge tubes with cap
- Aluminium foil
- Streptomyces coelicolor
- 32P-labelled orthophosphate (PerkinElmer, catalog number: NEX053H001MC )
Note: Use of radioactive materials requires institutional authorization. - Serine hydroxamate (Sigma-Aldrich, catalog number: S4503 )
- MilliQ quality water (dH2O)
- Formic acid (Thermo Fisher Scientific, Fisher Scientific, catalog number: A118P100 )
- 100% ethanol at room temperature
- Potassium phosphate monobasic (KH2PO4) (Sigma-Aldrich, catalog number: 60218 )
- 3-morpholinopropane-1-sulfonic acid (MOPS) (AMRESCO, catalog number: 0670 )
- Sucrose (Thermo Fisher Scientific, Fisher Scientific, catalog number: BP220-1 )
- Magnesium sulfate heptahydrate (AMRESCO, catalog number: 0662 )
- Dextrose/glucose (VWR, catalog number: BDH9230 )
- BactoTM yeast extract (BD, catalog number: 212750 )
- BactoTM peptone (BD, catalog number: 211677 )
- BactoTM tryptone (BD, catalog number: 211705 )
- Potassium phosphate dibasic (K2HPO4) (Sigma-Aldrich, catalog number: P3786 )
- Casamino acids (BD, catalog number: 228830 )
- L-histidine monohydrochloride monohydrate (Sigma-Aldrich, catalog number: H8125 )
- L-tryptophan (Sigma-Aldrich, catalog number: T0254 )
- L-tyrosine (Sigma-Aldrich, catalog number: T3754 )
- L-proline (Sigma-Aldrich, catalog number: P0380 )
- Springs (stainless) for dispersing the mycelia (e.g., Grainger, catalog number: 1NCH7 ) (specific to dispersal of S. coelicolor)
- Dry ice
- Guanosine monophosphate (Sigma-Aldrich, catalog number: G8377 ) (optional)
- Guanosine triphosphate (Sigma-Aldrich, catalog number: G8877 ) (optional)
- Modified MOPS minimal media (for growing S. coelicolor, see Recipes)
- 1.5 M KH2PO4 (pH 3.4) (see Recipes)
- 13 M formic acid (see Recipes)
- 100 mM serine hydroxamate (see Recipes)
- ISP1 medium (for growing S. coelicolor, see Recipes)
Equipment
- 50 ml glass Erlenmeyer flasks
- TLC chamber with lid (Sigma-Aldrich, catalog number: Z126195 )
- Phosphorimager (GE Healthcare)
- Phosphor screens with storage cassettes (GE Healthcare)
- Refrigerated microcentrifuge with speed up to 16,000 x g and rotor that fits 1.5 ml centrifuge tubes
- Shaker-incubator with temperature control
Software
- ImageQuant software
Procedure
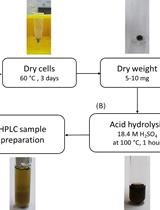
- Setting up the culture (Figure 1)
- Grow Streptomyces coelicolor in 5 ml ISP1 media in sterile 50 ml glass flask loosely covered with aluminium foil in shaking incubator (180 rpm) at 28 °C overnight.
Notes:- The incubation temperature is specific to S. coelicolor and may need to be adjusted for other bacterial species.
- Include springs in the growth media to disperse the mycelia and reduce clumping.
- The incubation temperature is specific to S. coelicolor and may need to be adjusted for other bacterial species.
- Add 150 μl of this culture to 600 μl modified MOPS minimal media in sterile 1.5 ml tubes.
- Disperse the mycelia well by vortexing and pipetting and transfer 200 μl to two sterile 1.5 ml tubes.
- From this step forward, handle samples behind a radiation shield and follow appropriate practices for handling and disposal of radioactive materials. Only certified personnel authorized by the radiation safety office of their respective institutions should handle the radioactive materials.
- To both 200 μl culture samples, add 1.7 μl of 32P-labelled orthophosphate (total 30 µCi) and grow the culture in the shaker incubator for 6 h (OD600 0.4-0.6) at 28 °C.
- Add 75 μl of 100 mM serine hydroxamate to one of the tubes and add 75 μl of sterile dH2O to the other tube.
- Mix well and incubate at 28 °C in the shaker incubator for 15 min.
- Centrifuge the samples at 10,000 x g for 5 min at 4 °C, discard the supernatant, add 25 μl of 13 M formic acid to the pellet and resuspend the pellet by vortexing.
Note: Use of ice-cold formic acid will stop the metabolic activity rapidly. If possible, estimate the weight of the pellet by subtracting the weight of the empty tube from the weight of the tube with the pellet before adding formic acid and adjust the amount of formic acid used to resuspend the pellets if needed. Discard the supernatant immediately, otherwise the pellet will loosen. - For cell lysis, incubate on dry ice for 1 min and on ethanol bath at room temperature for 1 min.
- Repeat step 9 four times.
- Incubate the samples on ice for 30 min.
- Vortex the samples well and centrifuge at 12,000 x g for 10-15 min at 4 °C.
- Store the supernatant in 1.5 ml tubes at -20 °C.

Figure 1. Setting up the culture
- Grow Streptomyces coelicolor in 5 ml ISP1 media in sterile 50 ml glass flask loosely covered with aluminium foil in shaking incubator (180 rpm) at 28 °C overnight.
- Setting up thin layer chromatography (Figure 2)
- Pour 1.5 M KH2PO4 (pH 3.4) buffer into a TLC chamber such that it forms a thin layer (~0.5 cm) at the bottom (50 ml used here) and close the lid in order for the buffer vapors to saturate the chamber (at least 30 min).
Note: Allow sufficient time for the KH2PO4 buffer vapors to saturate the TLC chamber before inserting the PEI-cellulose plate. This process may be facilitated by first cutting filter papers to a size smaller than that of the PEI-cellulose plates and immersing them in the 1.5 M KH2PO4 buffer. Place the soaked paper against the sides of the TLC chamber to let the vapors diffuse evenly throughout the chamber. Saturating the chamber will prevent the mobile phase from evaporating as it migrates up the plate. - Using a pencil, draw a line approximately 1 cm from the bottom of the PEI-cellulose plate and make fine dots on it approximately 2 cm apart to mark spots for sample loading.
Notes: - If a smaller TLC chamber is used, cut the PEI-cellulose plates into an appropriate size so that it fits inside the chamber with the lid closed. A covered beaker can substitute for a conventional TLC chamber.
- Do not poke the brittle PEI cellulose plates while drawing the baseline and dots. Do not use ink-pen, as the dye may migrate with the mobile phase. Avoid touching the surface of the PEI cellulose plate.
- Use an amount of buffer that is just sufficient to immerse the bottom of the PEI-cellulose plate below the drawn baseline; the buffer should not touch the sample application line.
- Thaw samples (item 13 above) and spot 1 μl of the sample onto the fine dots on the baseline and air dry for 2-4 min. Note down the sample spotted at each point.
Note: For less concentrated samples and to get a denser spot of the nucleotide species, add increments of 1 μl to the same spot with intermittent air drying. Allowing each aliquot to dry before applying a second aliquot will prevent the diffusion of the sample that would occur if a greater volume were added at once. - Place PEI-cellulose plate in the chamber, which should be placed on a level surface such that the base of the TLC plate is evenly immersed in the buffer.
- Close the lid and allow the mobile phase buffer to migrate until it is near (but not above) the top of the plate (about 3 h).
- Take out the plate, mark the top of the buffer front with a pencil to identify the distance migrated by the mobile phase, then air dry the plate for 15 min or longer.
- Place the plate in a clean cassette exposing the radioactive surface to a phosphor screen for 24 h or more depending upon the intensity of the 32P signal.
- Scan the exposed phosphor screen on a phosphorimager and save the image.

Figure 2. Setting up TLC
- Pour 1.5 M KH2PO4 (pH 3.4) buffer into a TLC chamber such that it forms a thin layer (~0.5 cm) at the bottom (50 ml used here) and close the lid in order for the buffer vapors to saturate the chamber (at least 30 min).
Data analysis
- Calculate the relative distance of migration (Retention Factor, Rf) of the different phosphorylated nucleoside species from Rf = (Distance travelled by nucleotide species)/(distance travelled by the mobile phase), with both distances measured relative to the baseline drawn on the TLC plate (origin; Figure 3). Rf values obtained under identical conditions may be compared to previously published results for identification of compounds (Calderón-Flores et al., 2005; Mechold et al., 2013). The more highly phosphorylated species are adsorbed more strongly to the PEI on the TLC plate and will migrate a shorter distance. Repeat the experiment at least twice using independent cultures to verify the data.
Notes: - The actual distance migrated by the nucleotides depends on the distance of migration of the mobile phase, requiring calculation of the Rf value for comparison purposes.
- For verification of sample identity, unlabeled nucleotides may be spotted on the plate and their migration identified by exposure to UV-light; the plate is fluorescent under UV-light, and the nucleotides will absorb the light, causing the appearance of a shadow.
- Example data analysis and calculation of Rf:
Distance traveled by mobile phase = 12.2 cm; distance migrated by GTP = 6.5 cm; distance migrated by ppGpp = 2.9 cm; distance migrated by pppGpp = 1.7 cm.
For GTP, Rf = 6.5 cm/12.2 cm = 0.53
For ppGpp, Rf = 2.9 cm/12.2 cm = 0.23
For pppGpp, Rf = 1.7 cm/12.2 cm = 0.13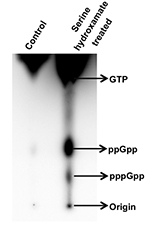
Figure 3. Thin layer chromatography of nucleotides (phosphorylated guanosines) extracted from exponential phase cultures of S. coelicolor treated without (first lane) and with 30 mM serine hydroxamate (second lane) for 15 min. The figure is modified from Figure 10A from Sivapragasam and Grove (2016). - Quantitate the density of spots corresponding to different nucleotide species using ImageQuant software. To compare relative levels of ppGpp and pppGpp between different samples, spot intensity of a nucleotide species may be divided by the combined intensity of both ppGpp and pppGpp after correcting the spot intensity for number of phosphates per nucleotide.
Example data analysis and calculation of relative levels of ppGpp and pppGpp: - Spot density of ppGpp after correcting for number of phosphates: Density of ppGpp obtained from ImageQuant (volume/area) divided by 4 (number of phosphates) = 131.52/4 = 32.88
- Spot density of pppGpp after correcting for number of phosphates: Density of pppGpp obtained from ImageQuant (volume/area)/5 (number of phosphates) = 23.32/5 = 4.66
- Total corrected spot density of ppGpp and pppGpp: 32.88 + 4.66
- Relative level of ppGpp: Corrected spot density of ppGpp/total corrected spot density of ppGpp and pppGpp = 32.88/(32.88 + 4.66) x 100% = 87.6%
- Relative level of pppGpp: Corrected spot density of pppGpp/total corrected spot density of ppGpp and pppGpp = 4.66/(32.88 + 4.66) x 100% = 12.4%
Notes
- Since it can be challenging to keep S. coelicolor mycelia dispersed during growth, collecting several samples from each culture condition is advised to increase the chances of collecting a comparable number of cells for each condition.
- It should be noted that serine hydroxamate does not induce stringent response in all bacterial species.
Recipes
- Modified MOPS minimal media (for growing S. coelicolor)
100 mM MOPS
10% sucrose
1% glucose
2 mM MgSO4
0.5% yeast extract
0.2% peptone
0.15% casamino acids
30 mM K2HPO4 (as in complete medium)
Add all the above ingredients in 800 ml of deionized water. Autoclave the buffer
50 mg of the amino acids histidine, proline, tyrosine and tryptophan (of each amino acid) is added to 100 ml of deionized water. Mix and slightly heat until all the powder dissolves. Filter the solution and add to the buffer prepared above. Make up the volume to 1 L using autoclaved deionized water. - 1.5 M KH2PO4 (pH 3.4)
20.4 g of KH2PO4 in 60 ml of dH2O
Adjust the pH to 3.4 using HCl
Make up the volume to 100 ml with dH2O - 13 M formic acid
Slowly add 2.787 ml of formic acid stock solution (88%) to 1.25 ml of dH2O
Make up the volume to 5 ml using dH2O
Mix well and freeze the solution at -20 °C - 100 mM serine hydroxamate
Add 12.11 mg of the powder to 1 ml of dH2O and vortex
Filter sterilize and store at -20 °C - ISP1 medium (for growing S. coelicolor)
Add 5 g tryptone and 3 g yeast extract to 1 L of dH2O
Autoclave
Acknowledgments
This work was supported by the National Science Foundation (MCB-1515349 to A.G.). We would like to thank Nabanita Bhattacharyya for assisting with the photography and all Grove lab members for their suggestions and helpful discussions. This protocol was derived from (Sivapragasam and Grove, 2016) originally published in Molecular Microbiology.
References
- Calderon-Flores, A., Du Pont, G., Huerta-Saquero, A., Merchant-Larios, H., Servin-Gonzalez, L. and Duran, S. (2005). The stringent response is required for amino acid and nitrate utilization, nod factor regulation, nodulation, and nitrogen fixation in Rhizobium etli. J Bacteriol 187(15): 5075-5083.
- Mechold, U., Potrykus, K., Murphy, H., Murakami, K. S. and Cashel, M. (2013). Differential regulation by ppGpp versus pppGpp in Escherichia coli. Nucleic Acids Res 41(12): 6175-6189.
- Sivapragasam, S. and Grove, A. (2016). Streptomyces coelicolor XdhR is a direct target of (p)ppGpp that controls expression of genes encoding xanthine dehydrogenase to promote purine salvage. Mol Microbiol 100(4): 701-718.
- Strauch, E., Takano, E., Baylis, H. A. and Bibb, M. J. (1991). The stringent response in Streptomyces coelicolor A3(2). Mol Microbiol 5(2): 289-298.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Sivapragasam, S. and Grove, A. (2016). Determination of (p)ppGpp Levels During Stringent Response in Streptomyces coelicolor by Thin Layer Chromatography. Bio-protocol 6(21): e1995. DOI: 10.21769/BioProtoc.1995.
Category
Microbiology > Microbial biochemistry > Other compound
Microbiology > Microbial physiology > Stress response
Biochemistry > Other compound > (p)ppGpp
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.