- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Ribosome Footprinting Protocol for Plants




Published: Vol 6, Iss 21, Nov 5, 2016 DOI: 10.21769/BioProtoc.1985 Views: 15485
Reviewed by: Samik BhattacharyaRenate WeizbauerBaohua Li
Original research article
The authors used this protocol in:

Oct 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Ribosome footprinting, or Ribo-seq, has revolutionized the studies of translation. It was originally developed for yeast and mammalian cells in culture (Ingolia et al., 2009). Herein, we describe a plant-optimized hands-on ribosome footprinting protocol derived from previously published procedures of polysome isolation (Ingolia et al., 2009; Mustroph et al., 2009) and ribosome footprinting (Ingolia et al., 2009; Ingolia et al., 2013). With this protocol, we have been able to successfully isolate and analyze high-quality ribosomal footprints from different stages of in vitro grown Arabidopsis thaliana plants (dark-grown seedlings [Merchante et al., 2015] and 13-day-old plantlets in plates and plants grown in liquid culture [unpublished results]).
Background
The central role of translation in modulating gene activity has long been recognized, yet the systematic exploration of quantitative changes in translation at a genome-wide scale in response to a specific stimulus has only recently become technically feasible. The ribosome footprinting technology (often known as the Ribo-seq), developed originally for yeast and mammalian cells in culture, has revolutionized the studies of translation regulation and gene expression, as it allows to determine the exact positions of the ribosomes at a genome-wide scale and at a single-codon resolution (Ingolia et al., 2009).
Prior to the development of Ribo-seq, the most common methods employed to study translation regulation in plants were the isolation of polysomal RNA via sucrose gradient centrifugation or translating ribosome affinity purification (TRAP) followed by Northern blotting, qRT-PCR, microarrays, or RNA-seq. The first method, known as polysome profiling, relies on resolving distinct polysomal fractions on a sucrose gradient via ultracentrifugation (Branco-Price et al., 2008; Missra and von Arnim, 2014; Li et al., 2015). By comparing different plant growth conditions or mutants, one could infer the changes in the rates of translation from observing a shift in the distribution of mRNAs between polysomal fractions. For example, if a transcript becomes more abundant in the monosomal fraction with the concomitant decrease in the higher order polysomes, the translation of this mRNA is considered as down-regulated. The key limitation of this technique, however, is its low resolution of higher-order polysomes (and thus mild quantitative changes in translation are often missed) and the inability to differentiate between polysomal RNAs that undergo active translation versus are loaded with arrested ribosomes (for example, those ‘stuck’ in the upstream open reading frames of a transcript). The second polysomal RNA isolation technique, TRAP, is based on the stable expression of an epitope-tagged ribosomal protein followed by the immunoprecipitation of entire ribosomes along with their associated mRNAs (Zanneti et al., 2005; Reynoso et al., 2015). While this latter method accommodates both global and tissue-specific studies of translation (achieved by driving the expression of a tagged ribosomal protein in a ubiquitous versus tissue-specific manner), its use is limited to transformable species where transgenic lines can be generated. Furthermore, transcriptomic analysis of TRAP samples per se does not provide a quantitative measure of translation (unless coupled with Ribo-seq [Juntawong et al., 2014]), as any mRNA with one or more ribosomes bound to it will be purified by TRAP. Also, since TRAP relies on epitope-tagging and the tag may interfere with the function of the ribosome, the regulation of translation of some mRNAs may be disrupted in the TRAP transgenic lines, e.g., due to a reduced ability of the tagged ribosome to associate with specific proteins at certain stages of translation. Another limitation of TRAP is that it typically uses a specific redundant isoform of a ribosomal protein for tagging, such as RPL18, and thus likely purifies only a subset of ribosomes that carry just that RPL18 variant. Given that there are multiple RPL18-like proteins in plant genomes, using one specific ribosomal protein isoform for tagging misses the ribosomes that utilize an alternative RPL18 isoform.
The method of choice for our studies, the Ribo-seq, does not involve transgenic line generation nor affinity purification, thus avoiding many of the limitations of the aforementioned earlier techniques. Most importantly, the single-codon resolution of the ribosome footprinting technology allows researchers to map the ribosomes on the mRNAs and thus clearly distinguish between the transcripts harboring productive ribosomes translating the main genic open reading frames versus transcripts associated with non-productive ribosomes arrested in the 5’UTRs. Not only does this method offer a snapshot of a whole-genome view of ribosomal distribution at an unprecedented resolution, it also enables the true quantitative measure of translational efficiency of every expressed gene in the genome by correlating the Ribo-seq data with the transcriptional information obtained via RNA-seq. Nonetheless, even the Ribo-Seq has its own drawbacks, as it cannot discriminate between mRNA subpopulations with different translation efficiencies, giving an average translation efficiency readout for each expressed gene.
Herein, we provide a plant-optimized Ribo-seq protocol that enables the study of translation regulation through the isolation of high-quality ribosomal footprints from different developmental stages of in vitro grown Arabidopsis thaliana seedlings. It describes step by step how to pellet and digest polysomes, isolate monosomes, extract the mRNA footprints, and generate sequencing libraries for the Illumina platform. The protocol also describes the preparation of parallel RNA-seq libraries to account for transcriptional regulation. We conclude the description of our method with a brief summary of how to analyze the sequencing results.
Materials and Reagents
- Materials
- Miracloth
- 30 ml NalgeneTM High-Speed polycarbonate centrifuge tubes (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 3118-0030 ) (or equivalent) to centrifuge the plant extracts
- 5 ml polypropylene thin-wall ultracentrifuge tubes (Beckman Coulter, catalog number: 326819 )
- 10 ml ultracentrifuge 14 x 89 mm tubes with isopycnic caps (BioComp Instruments, catalog number: 105-914A )
- Fine scale
- 10 ml syringe
- Cannula to be attached to the syringe (Thomas Scientific, catalog number: 1193G13 )
- Pre-sterilized, RNase-free 5 ml, 1 ml, 200 µl and 10 µl micropipette tips
- Pre-sterilized, RNase-free 2 ml and 1.5 ml microcentrifuge tubes
- 15 and 50 ml Falcon tubes
- Stoppers to secure the lids of 1.5 ml tubes
- Razor blades
- Dynabeads mRNA Purification Kit (Thermo Fisher Scientific, AmbionTM, catalog number: 610-06 )
- 2 ml microcentrifuge tubes
- 1.5 ml non-stick, RNase-free microtubes (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: AM12450 )
- MyOneTM Streptavidin C1 DynaBeads® (Thermo Fisher Scientific, InvitrogenTM, catalog number: 65601 )
- 200 μl PCR strip tubes
- Miracloth
- Reagents
- Liquid nitrogen
- RNase-free, sterile, MilliQ water
- Tris base
- Sucrose
- Potassium chloride (KCl)
- Sodium chloride (NaCl)
- Magnesium chloride (MgCl2)
- Ethyleneglycol-bis(2-aminoethylether)-N,N,N’,N-tetraacetic acid (EGTA)
- Ethylene diamintetracetic acid (EDTA)
- Dithiothreitol (DTT)
- Brij-35 [Polyoxyethylene(23)lauryl ether]
- Triton X-100
- Igepal CA 630 (Octylphenyl-polyethylene glycol)
- Tween 20 (Polyoxyethylene sorbitan monolaurate 20)
- Cycloheximide (see Note 1)
- Chloramphenicol (see Note 1)
- Lithium chloride (LiCl)
- Sodium acetate (NaOAc)
- Sodium bicarbonate (NaHCO3)
- Sodium carbonate (Na2CO3)
- Sodium dodecyl sulfate (SDS)
- Sodium hydroxide (NaOH)
- Trisodium citrate (Na3C6H5O7)
- Dimethylsulfoxide (DMSO), PCR grade
- Ethanol, molecular biology grade
- Water-saturated, acid phenol, molecular biology grade (see Note 2)
- Chloroform, molecular biology grade (see Note 2)
- Isoamyalcohol
- Isopropanol, molecular biology grade
- PEG8000
- 15 mg/ml GlycoBlueTM (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9515 )
- 10 bp DNA ladder (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10821015 )
- 2x denaturing sample buffer with dye (Thermo Fisher Scientific, InvitrogenTM, catalog number: LC6876 ) (see Note 3)
- SYBR® Gold, 10,000x in DMSO (Thermo Fisher Scientific, InvitrogenTM, catalog number: S11494 ) (see Note 4)
- dNTP mix, 10 mM (Thermo Fisher Scientific, InvitrogenTM, catalog number: 18427-013 )
- OmniPur® polyethylene glycol 8000 (EMD Millipore, Calbiochem®, catalog number: 6510 )
- Universal miRNA cloning linker (New England Biolabs, catalog number: S1315S )
- Boric acid
- Acetic acid
- 12-well 15% polyacrylamide TBE-Urea gels (Bio-Rad Laboratories, catalog number: 4566055 ) (see Note 5)
- 30% acrylamide/bisacrylamide (29:1) (Bio-Rad Laboratories, catalog number: 161-0156 ) (see Note 5)
- Ammonium persulfate (APS)
- TEMED (N,N,N’,N’-Tetramethylethane-1,2-diamine) (Sigma-Aldrich, catalog number: T7024 )
- Glycerol
- Bromophenol blue
- Liquid nitrogen
- Enzymes
- SUPERase-InTM RNase inhibitor (Thermo Fisher Scientific, AmbionTM, catalog number: AM2694 )
- TURBOTM DNase (2 U/μl) (Thermo Fisher Scientific, AmbionTM, catalog number: AM2238 )
- RNase I (100 U/μl) (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM2294 )
- SuperScript® III reverse transcriptase (Thermo Fisher Scientific, InvitrogenTM, catalog number: 18080-093 )
- T4 polynucleotide kinase, T4 PNK4 (New England Biolabs, catalog number: M0201 ), supplied with 10x T4 PNK buffer (New England Biolabs, catalog number: B0201 ) (see Note 6)
- T4 RNA ligase 2, truncated (New England Biolabs, catalog number: M0242 ), supplied with PEG 8000 50% and 10x T4 Rnl2 buffer
- Phusion high-fidelity DNA polymerase (2 U/μl) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: F-530S )
- CircLigaseTM ssDNA ligase (100 U/μl) (Epicentre, catalog number: CL4115K )
- SUPERase-InTM RNase inhibitor (Thermo Fisher Scientific, AmbionTM, catalog number: AM2694 )
- Oligonucleotides
- Reverse transcription primer for split-adapter circularization (see Note 7)
NI-NI-9: [P]AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGC[SC18]CACTCA[SC18]TTCAGACGTGTGCTCTTCCGATCTATTGATGGTGCCTACAG (Ingolia et al., 2009) - Size marker oligos (Ingolia et al., 2009) (see Note 8)
oNTI265: rArUrGrUrArCrArCrGrGrArGrUrCrGrArGrCrUrCrArArCrCrCrGrCrArArCrGrCrGrA
oNTI268: rArUrGrUrArCrArCrGrGrArGrArCrCrCrGrCrArArCrGrCrGrA - rRNA subtraction oligos (all the oligos here have a 5’ TEG-linked biotin and were obtained from IDT),These oligos are based on the most abundant rRNA sequences and transposons that were found using 3-day-old Arabidopsis seedlings and the ribosome footprinting protocol described herein (see Note 9)
rRNABio1: 5’-gataaccgtagtaattctagag-3’
rRNABio2&4: 5’-TGATTCATGATAACTCGACGGACGACGCGGATTACGGTGGCGGC-3’
rRNABio5&3: 5’-GTCGCTGCCGTGATCGTGGTCTCCATCGAGTCTTTGAACGCAAG-3’
Bio-Tranpos: 5’-GAGGGATGCAACACGAGGAGTTCCCGGGAGGTCA-3’
Bio-5S: 5’-AAGCCTTCTGGCCGAGGGCACGTCTGCCTGGGTGTCACAA-3’
Bio-18S: 5’-AAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTG-3’ - Amplification primers
NI-NI-2: 5’-AATGATACGGCGACCACCGAGATCTACAC-3’ (Ingolia et al., 2009)
NI-NI-3: 5’-CAAGCAGAAGACGGCATACGAGATAGTCGTGTGACTGGAGTTCAGACGTGTGCTCTTCCG-3’ (Ingolia et al., 2009)
Alternative indexed primers
Index 1:
5’-CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCAGACGTGTGCTCTTCCG-3’
Index 2:
5’-CAAGCAGAAGACGGCATACGAGATACATCGGTGACTGGAGTTCAGACGTGTGCTCTTCCG-3’
Index 3:
5’-CAAGCAGAAGACGGCATACGAGATGCCTAAGTGACTGGAGTTCAGACGTGTGCTCTTCCG-3’
Index 4:
5’-CAAGCAGAAGACGGCATACGAGATTGGTCAGTGACTGGAGTTCAGACGTGTGCTCTTCCG-3’
Index 5:
5’-CAAGCAGAAGACGGCATACGAGATCACTGTGTGACTGGAGTTCAGACGTGTGCTCTTCCG-3’
Index 6:
5’-CAAGCAGAAGACGGCATACGAGATATTGGCGTGACTGGAGTTCAGACGTGTGCTCTTCCG-3’
Index 7:
5’-CAAGCAGAAGACGGCATACGAGATGATCTGGTGACTGGAGTTCAGACGTGTGCTCTTCCG-3’
- Reverse transcription primer for split-adapter circularization (see Note 7)
- Solutions
All solutions should be prepared with RNase-free, MilliQ water. Unless otherwise stated, all of them must be sterilized by autoclaving for at least 20 min and stored at room temperature.- 1 M Tris-HCl, pH 7.0 (see Note 10)
- 1 M Tris-HCl, pH 8.0 (see Note 10)
- 1 M Tris-HCl, pH 9.0 (see Note 10)
- 1 M sucrose, prepared from Molecular Biology grade sucrose (autoclave for 10 min)
- 1 M KCl
- 5 M NaCl
- 1 M MgCl2
- 0.5 M EGTA, pH 8.0 (see Note 11)
- 0.5 M EDTA, pH 8.0 (see Note 11)
- 2 mM EDTA, 100 mM Na2CO3
- 2 mM EDTA, 100 mM NaHCO3
- 3 M NaOAc, pH 5.5
- 10% SDS (w/v)
- 1 M NaOH (no sterilization required)
- 4 M LiCl (filter-sterilize, do not autoclave)
- 1 M DTT (do not autoclave; aliquot and store at -20 °C)
- 100 mg/ml cycloheximide, prepared in DMSO (filter-sterilize; aliquot and store at -20 °C)
- 50 mg/ml chloramphenicol, prepared in ethanol (filter-sterilize; aliquot and store at -20 °C)
- Detergent mix 20% (w/v or v/v) of each of four detergents in water (Brij-35, Triton X-100, Igepal CA 630 and Tween 20)
- 50% PEG8000 (w/v), prepared by combining dry PEG with water in a 50 ml conical and mixing with gentle shaking overnight. No sterilization is required. PEG is very hygroscopic; so start adding PEG to less than half of the final volume of MilliQ water. Then, after it is dissolved, add more water if needed. PEG used for ligation (step C3) should not be more than 1 month old.
- 10% APS (w/v), Filter-sterilize and aliquot in small volumes (e.g., 500 µl). Store at -20 °C. Aliquots can be thawed and re-frozen several times.
- 1 M Tris-HCl, pH 7.0 (see Note 10)
- Buffers (see Recipes)
- Polysome extraction buffer (PEB)
- Sucrose cushion A (SCA)
- Sucrose gradients solutions
- Polysome digestion buffer (PDB)
- Sucrose cushion B (SCB)
- Polysome resuspension buffer (PRB)
- Total RNA extraction buffer (TREB)
- Alkaline fragmentation buffer (2x)
- Alkaline fragmentation stop/precipitation solution
- TAE (Tris/acetate/EDTA buffer) (50x)
- TBE (Tris/borate/EDTA buffer) buffer (5x)
- RNA gel extraction buffer (GEB)
- DNA gel extraction buffer (STE)
- SSC (20x)
- Subtraction bind/wash buffer (2x)
- 8% non-denaturing polyacrylamide gel (12 ml)
- Non-denaturing loading dye (6x)
- Polysome extraction buffer (PEB)
Equipment
- 7-9 cm diameter porcelain mortar and pestle
- Small (30-50 ml) glass beakers
- Refrigerated Beckman Avanti J-25 centrifuge and Beckman JA17 rotor (or equivalent)
- Ultracentrifuge Beckman L8-70M (or equivalent) with swinging bucket rotors for polysome pelleting (Beckman Coulter, model: SW55Ti ) and sucrose gradient centrifugation (Beckman Coulter, model: SW41Ti or Thermo Fisher Scientific, Thermo ScientificTM, model: TH-641 ).
- Automatic P1000, P200 and P10 micropipettes
- Refrigerated tabletop microcentrifuge
- Orbital shaker
- Gradient Master Station, with both the gradient maker and the fractionation station (BioComp Instruments)
- Continuous UV light monitor (Bio-Rad Laboratories, model: EM-1 Econo UV Monitor )
- Fractionator (Gilson, model: FC203B )
- Nanodrop (or another spectrophotometer to quantify nucleic acid concentrations in small volumes of RNA)
- Nutator shaker
- Thermoblock
- Fume hood
- Mini-PROTEAN tetra cell polyacrylamide gel box (Bio-Rad Laboratories, catalog number: 165-8004 ) or equivalent and electrophoresis power supply
- UV transilluminator
- Vortex
- DynaMagTM-2 magnetic separation rack (Thermo Fisher Scientific, catalog number: 12321D )
- Thermocycler
- 2100 BioAnalyzer (Agilent Technologies, catalog number: G2940CA )
- HiSeq2000 (Illumina)
Procedure
The wet-lab protocol is divided into A, B, and C steps (see Figure 1 for an overview). A steps regard to the preparation of the ribosome footprints. B steps refer to the total mRNA isolation procedure. C steps represent the part of the protocol where the footprints and mRNA fragments are processed together to create the sequencing libraries. The order in which A and B steps are performed is up to the researcher.
Tissues need to be flash-frozen after the desired treatment, ground to a very fine powder in liquid nitrogen, and then stored in -80 °C until use. 0.5 g of this pulverized tissue is required for the ribosome footprints and 0.5-1 g are needed to prepare the mRNA fragments. It is good practice to first isolate polysomes without including the digestion step to ensure that intact polysomes are being isolated before proceeding to the preparation of the footprint libraries. As with all RNA protocols, the tissues should not be allowed to thaw in the absence of buffer at any moment and all materials and solutions need to be RNase free. All the work should be performed on ice, unless otherwise stated.
These protocols allow for the generation of ready-to-sequence Ribo-seq and RNA-seq libraries from frozen ground tissues in 9 days. The entire protocol is organized in working days. At the end of the protocol, we include a Data analysis section that describes the pipeline that leads to the identification of genes regulated at the level of translation by the treatment or mutation of interest.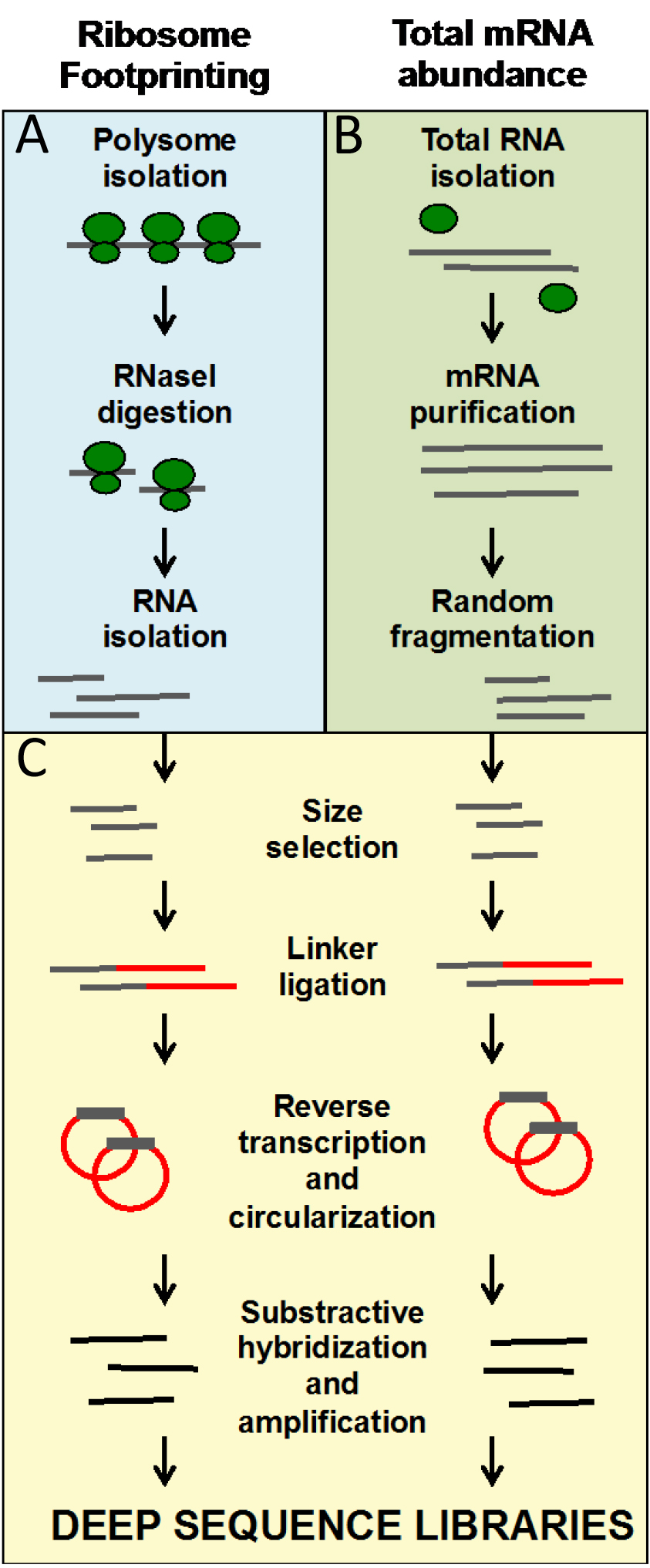
Figure 1. Overview of the ribosome footprinting procedure. A. Blue panel: steps that are specific to ribosome footprinting; B. Green panel: steps specific to mRNA library preparation; C. Yellow panel: steps that are common to both types of libraries. A, B and C steps are also marked in the written protocol.
- Steps: Ribosome footprinting
Figure 2 shows a flow chart of the procedure comprising the A steps.
Figure 2. Overview of the A steps of the protocol
Note: Day 1 is a long day, around 14-18 h, dependent on the number of samples to be processed and the skills of the worker.
A1. Polysome isolation
- Switch ‘on’ the Beckman Avanti J-25 centrifuge and the L8-70M ultracentrifuge to allow them to cool down to 4 °C. Pre-chill the JA17 and SW55Ti rotors as well.
- Pre-chill on ice a clean porcelain mortar and pestle, a clean beaker with 3 layers of Miracloth (arranged as a funnel), and a Nalgene centrifuge tube per each sample to be analyzed.
- Place 0.5 g of pulverized frozen tissue in the pre-chilled mortar and add immediately 2 ml of ice-cold polysome extraction buffer (PEB). Mix the sample well with the pestle so that all the tissue is thawed in contact with the buffer.
- Let the sample stand on ice (with occasional gentle mixing with the pestle) for 10 min or until the rest of the samples are prepared.
- Take all the mixture with a 1 ml pipette or pour it into the beaker through the Miracloth filter to remove the biggest particles. Squeeze the filter to make sure that all of the supernatant is being collected in the beaker. Wash the mortar with 1 ml of new PEB, and pass it through the Miracloth. Squeeze again, and then, wash the Miracloth with 1 ml of clean PEB. Squeeze. Overall, a total of 4 ml of PEB is used for 0.5 g of tissue.
- Transfer the extract from the beaker to a Nalgene centrifuge tube making sure to collect as much extract as possible (see Note 12).
- Equalize the weight of the tubes with PEB before putting them in the Beckman Avanti J-25 centrifuge, so that the difference between opposite tubes is not bigger than 0.1 g.
- Centrifuge at 4 °C, at 7,728 x g, for 30 min.
- While the samples are spinning, prepare the sucrose cushion for polysome pelleting (steps A1.10 and A1.11).
- Pre-chill on ice the 5 ml ultracentrifuge tubes and buckets for the Beckman SW55Ti rotor.
- The sucrose cushion for polysome pelleting is made from sucrose cushion A (SCA) (see Recipes). SCA can be prepared in advance and stored in aliquots at -20 °C. If so, thaw on ice the amounts of SCA aliquots that are needed for the experiment taking into account the volume in which the aliquots were prepared (see Note 13) and that each ultracentrifuge tube will contain 0.5 ml of sucrose cushion and 4.5 ml of the plant extract (see Note 14).
- Once thawed, spin the SCA aliquots briefly in a tabletop microcentrifuge to collect the sucrose solution on the bottom of the tubes.
- Transfer the total needed volume of SCA with some excess to a new pre-chilled tube that fits the total volume of SCA that is required for the experiment (see Note 15). Homogenize the SCA solution by mixing carefully with the 1 ml pipette avoiding the formation of bubbles.
- To the SCA, add the DTT, cycloheximide and chloramphenicol to a final concentration of 5 mM, 50 μg/ml, and 50 μg/ml, respectively (see SCA in Recipes). Mix well with the pipette avoiding bubbles and transfer 0.5 ml of the cushion to each of the ultracentrifuge tubes.
- Once the centrifuge (step A1.8) is finished spinning, pour the supernatant into a clean, pre-chilled 15 ml tube (or any other type of tube larger than 4.5 ml) (see Note 16).
- Bring the volume of the extract to 4.5 ml with PEB and mix it carefully.
- Transfer the 4.5 ml extract to the ultracentrifuge tube on top of the sucrose cushion. A good way to do this is to take the extract with a 1 ml micropipette, place the tip perpendicular to the tube wall at the upper part of the tube, and let the extract slowly drip along the wall. This way the cushion will not be disturbed (Schematic representation in Figure 3A).
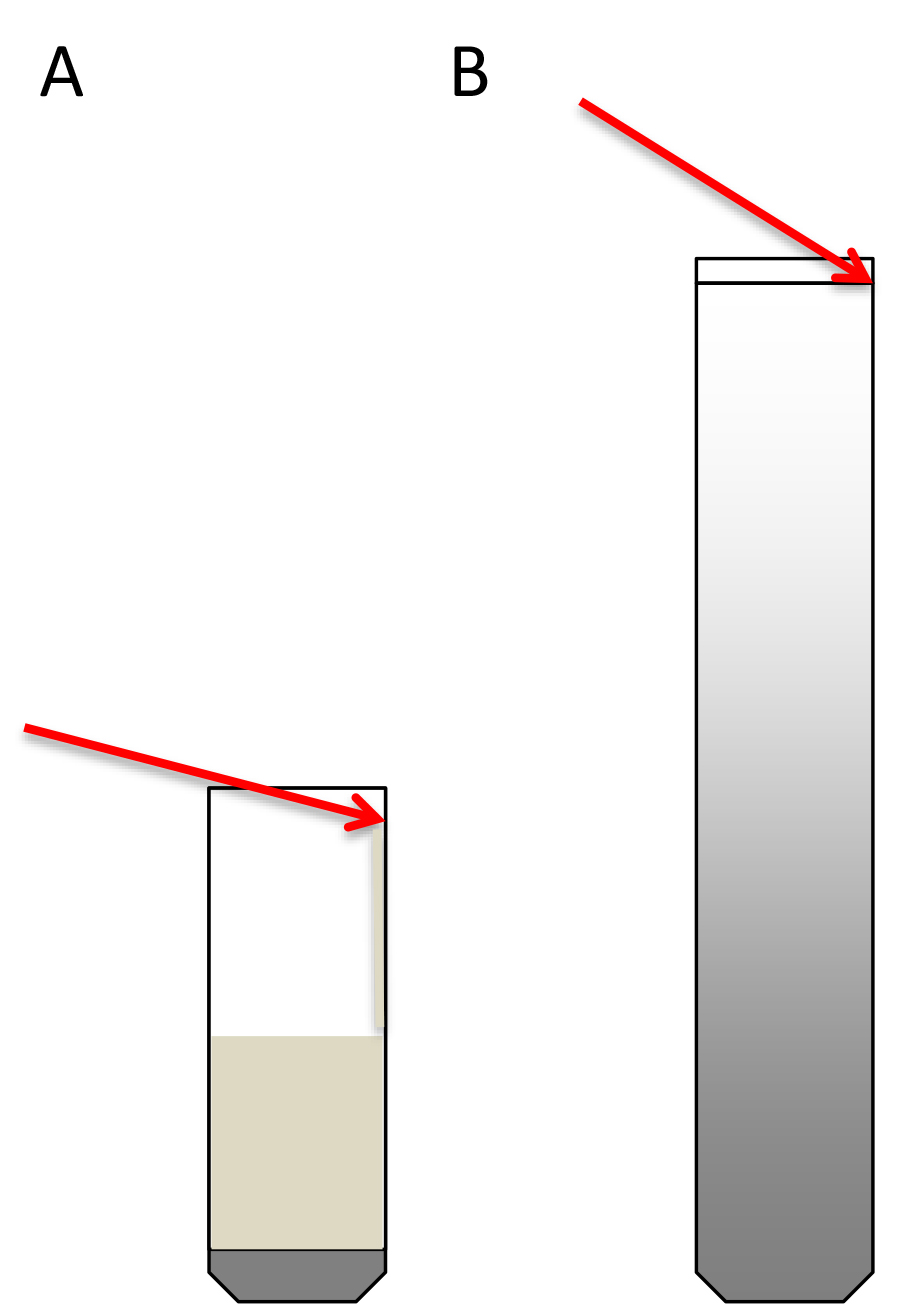
Figure 3. Schematic representation of how to load the sucrose cushions (A) and the sucrose gradients (B). Red arrows indicate the angle of the pipette. - Equalize the weight of opposite tubes carefully with PEB using fine scales.
- Place ultracentrifuge tubes into the pre-chilled buckets and those into the pre-chilled SW55Ti rotor, making sure that the number on the bucket coincides with the position in the rotor.
- Centrifuge at 4 °C at 256,677 x g, for 3.5 h.
- While the samples are spinning, prepare the sucrose gradients (steps A1.22-A1.29) (see Note 17).
- To make the sucrose gradients and to fractionate polysomes, use the Gradient Master Station from BioComp Instruments, Seton 7030 10 ml ultracentrifuge tubes, rubber caps with a small hole that close the ultracentrifuge tubes, marker block and syringe with a cannula from BioComp Instruments and follow manufacturer’s recommendations.
- Prepare at room temperature the 10% and 50% sucrose gradients solutions (see Recipes). Each gradient will need 5 ml of each sucrose solution. Prepare the amount needed for the experiment with some excess.
- Mark the 10 ml ultracentrifuge tubes according to the marker block with a permanent marker (see Note 18).
- Fill the marked tubes with the 10% sucrose solution up to the mark (5 ml approx.).
- With the syringe and the cannula underlay 50% sucrose solution below the 10% one to fill the tube entirely (another 5 ml approx.).
- Gently insert the rubber cap into the top of the tube at an angle, so that the hole in the cap enters the liquid last allowing the air to escape.
- Put the tubes in the gradient station’s magnet bucket and place them on the Gradient Master Station. Program the station in the gradient position, with a rotation angle of 81.5°, at speed 16, for 1:58 min.
- Once the gradients are prepared, place them carefully at 4 °C until they are used.
- When the centrifuge run (step A1.20) is close to finishing, prepare the polysome digestion buffer (PDB) (see Recipes) without adding the DNase and RNase. Also have ice-cold MilliQ water, 10 ml per sample, ready to wash the polysome pellets.
- Once the polysome pelleting ultracentrifuge is finished, remove all the supernatant and cushion by quickly and carefully pouring it down.
- Fill the tube with ice-cold MilliQ water and immediately pour it down. Repeat this washing step to remove all of the sucrose. The polysome pellet has a flat jelly-like appearance at the bottom of the tube (see Note 19).
- Briefly dry the pellets to remove most of the water by placing them upside down over clean absorbent paper for 30 sec-1 min.
- Resuspend the pellets in 500 μl of polysome digestion buffer (PDB) by pipetting carefully and transfer the samples to new clean 1.5 ml tubes.
A2. Polysome digestion and monosome pelleting
Polysome digestion is performed in a total volume of 4.5 ml. In our hands, this increase in the volume of digestion compared to other available footprinting protocols (Ingolia et al., 2009) significantly improves the quality of the obtained footprints. Digestion is performed in PDB. After digestion, the sample is pelleted again on a sucrose cushion before the fractionation step.When testing the protocol for the first time, it is recommended to keep one sample undigested to ensure that the polysomes are intact before the digestion step.
- Spin the 1.5 ml tubes with 500 μl of resuspended polysomes (step A1.34) for 30 sec at 20,800 x g and at 4 °C in a microfuge.
- Transfer the supernatant to a 15 ml Falcon tube pre-filled with 4 ml of PDB. Discard the pellet.
- Add 12 μl of TURBO DNase 2 U/μl and 10 μl of RNase I to each 4.5 ml sample for digestion (see Note 20).
- Incubate for 2 h at room temperature with gentle agitation in an orbital shaker.
- During the incubation, prepare the sucrose cushions for the next pelleting step. The cushions are prepared as described for the polysome pelleting step (steps A1.10-A1.14) but with sucrose cushion B instead (SCB) (see Recipes).
- When digestion time is over, add 7.5 μl of SUPERase-In RNase inhibitor to each 15 ml Falcon tube where polysome digestion is taking place and mix well by inversion. Immediately load the digested extracts onto the sucrose cushions as in step A1.17. Weight the tubes and equalize the weight of opposite tubes carefully with PDB using fine scales.
- Centrifuge, wash and dry the monosome pellets as before (steps A1.20 and A1.31-A1.33).
- Resuspend the pellets by pipetting carefully in 300 μl of polysome resuspension buffer (PRB) (see Recipes).
- Transfer to a new clean 1.5 ml tube. Spin briefly, and transfer the supernatant to a new tube. Discard the pellet.
A3. Polysome fractionation
At this step, the digested monosomal fraction is separated from the rest of the extract. To profile the polysomes, we recommend the use of the Gradient Master Station. Have the ultracentrifuge rotor and its centrifuge buckets pre-chilled at 4 °C. The ultracentrifuge should already be cold from the pelleting step, so keep it cold.
- Take the tubes with the sucrose density gradients (steps A1.22-A1.29) out of the fridge and place them on ice.
- Load the 300 μl of resuspended digested polysome pellets (step A2.9) on top of the sucrose density gradients. To do this, place the pipette tip at the wall of the tube, at the upper limit of the 10% sucrose meniscus. With the tip positioned there, slightly elevate the solution without breaking the surface tension and keep the tip in contact with the wall (Schematic representation in Figure 3B). Very carefully load the sample on top of the gradient. The extract should expand on top of the sucrose (this can be seen) without entering in it (see Note 21).
- Equalize the weight of all tubes very carefully with PRB using fine scales (be careful not to disturb the gradients).
- Carefully insert the tubes in the pre-chilled Sorvall TH-641 buckets and close them well. Put the buckets in the pre-chilled rotor making sure that the number on the bucket coincides with the position in the rotor.
- Spin the gradients in the ultracentrifuge at 4 °C at 234,479 x g for 2.5 h.
- 5-10 min before the centrifugation is finished, switch ‘on’ the Gradient Master Station and prepare it for the fractionation steps (follow manufacturer’s instructions).
- Retrieve the rotor buckets and keep the tubes on ice until fractionation.
- Fractionate the sucrose gradients on the gradient station at 0.2 mm/sec following manufacturer’s indications. Measure the A260 of the collected material using the continuous UV monitor to identify the 80S ribosome peak.
- Analyze the undigested sample first to ensure that it contains intact polysomes and to determine the approximate location of the 80S ribosome peak.
- Then, fractionate the digested sample, which should have a much larger 80S monosome peak than the undigested sample. The number of fractions depends on the researcher’s needs. Typically, collecting in 20 fractions allows the monosome peak to fall in a maximum of 2 fractions. Collected in this way, each fraction will contain an approximate volume of 0.5 ml. To collect the fractions, use 2 ml tubes.
- Keep the fractions(s) corresponding to the 80S peak and store the samples at -80 °C until the researcher is ready to isolate the footprints.
A comparison between undigested and digested polysome profiles is shown in Figure 4.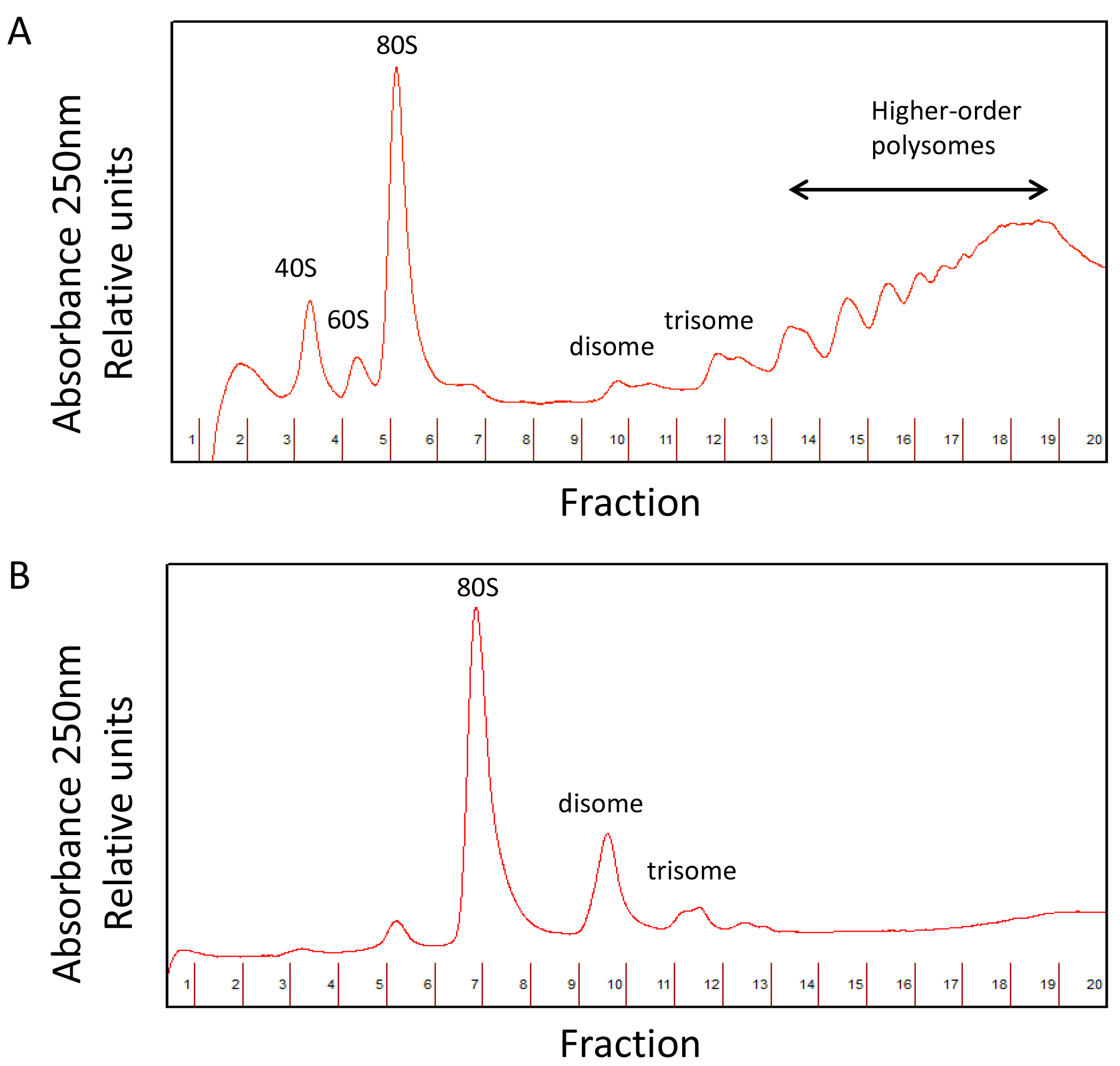
Figure 4. Undigested (A) and digested polysome (B) profiles
A4. Footprint fragment purification
Purify RNA from the monosome fraction using the SDS/phenol method. Work in a fume hood and use a lab coat, gloves and goggles to prevent skin damage if phenol spills.
- Thaw the monosome fractions (step A3.11) on ice. Spin briefly to collect the sample at the bottom of the tube.
- Add 10% SDS to the monosome fraction to a final concentration of 1% (w/v) (e.g., 50 µl of SDS are needed if the sample was fractionated into 20 fractions of 500 µl each), mix the sample by vortexing and place it at 65 °C. Secure the lids.
- Heat acid phenol-chloroform (5:1) to 65 °C in a fume hood.
- Add 1 volume of hot acid phenol-chloroform to the monosome fraction, mix well by vortexing and incubate for 5 min at 65 °C. Keep vortexing frequently while processing all of the samples (see Note 22).
- Place samples on ice for 5 min, and then spin at 20,800 x g in a tabletop microcentrifuge 2 min at room temperature.
- Transfer the upper aqueous phase (the lower phase if phenol-chloroform) to a new 1.5 ml tube (see Note 23).
- Add 1 volume of acid phenol-chloroform and keep it for 5 min at room temperature vortexing frequently.
- Spin for 2 min at room temperature at 20,800 x g in a tabletop microcentrifuge.
- Carefully transfer the aqueous phase to a new 1.5 ml tube and add 1 volume of chloroform-isoamylalcohol (24:1). Vortex for 1 min at room temperature.
- Spin for 1 min at room temperature at 20,800 x g in a tabletop microcentrifuge.
- Transfer the aqueous phase to a new pre-chilled 1.5 ml tube. If the digested polysomes were fractionated into 20 fractions of 0.5 ml, a recovery of 400 μl aqueous phase is expected.
- Precipitate RNA using GlycoBlue as a coprecipitant. To 400 μl RNA add 45 μl of 3 M NaOAc (pH 5.5), and 6.75 μl of GlycoBlue. Mix well by inversion and add 675 μl of ice-cold isopropanol. Mix well by inverting the tube.
- Incubate at least 30 min at -80 °C or dry ice, and centrifuge for 30 min at 20,800 x g at 4 °C in a tabletop microcentrifuge.
- Discard the supernatant by inverting the tube, spin briefly to collect the drops, remove them with the pipette, and air-dry the pellet for 1 min at room temperature.
- Resuspend the RNA in 10 μl of 10 mM Tris-HCl, pH 8.0.
- Keep the footprint RNA at -80 °C until the size selection step in gel (see step C1), which will be performed in parallel with the fragmented mRNA samples.
- Steps: total mRNA preparation
B1. Total RNA extraction
Multiple RNA purification protocols could be used at this step. The following is based on the Reuber and Ausubel RNA extraction protocol (Reuber and Ausubel, 1996) (see Note 24).
- Switch ‘on’ a Beckman Avanti J-25 centrifuge to cool it down to 4 °C. Pre-chill the polycarbonate Nalgene centrifuge tubes on ice and the JA-17 Beckman rotor in the refrigerator.
- Add 5 ml of ice-cold total RNA extraction buffer (TREB) (see Recipes) to the pre-chilled centrifuge tubes, keep them on ice and add 0.5-1 g of pulverized frozen tissue.
- Vortex and invert the tube to make sure that all the tissue is in contact and thawed in the buffer. Keep the extracts on ice until the remaining samples are processed.
- Add 1 volume of acid phenol (see Note 25), vortex to uniformly mix the phenol and extract. Keep the samples on ice vortexing frequently while the remaining samples are processed.
- Spin samples at 4 °C at 7,728 x g for 10 min.
- Transfer the upper aqueous phase (the bottom phase is phenol) to a new polycarbonate centrifuge tube prefilled with 5 ml of acid phenol, vortex, and spin the samples in the centrifuge as in step B1.5.
- Transfer the upper aqueous phase to a new polycarbonate centrifuge tube prefilled with 5 ml of chloroform, vortex, and spin the samples at 4 °C, 7,728 x g for 10 min.
- Collect the supernatant and divide it into 10 pre-chilled 2 ml microcentrifuge tubes with 500 µl extract in each (see Note 26).
- To each tube add 0.1 volumes of 3 M NaOAc, pH 5.5 mix, and add 2.5 volumes of 100% ethanol (see Note 27).
- Mix samples by inversion. Place samples on ice for 5-10 min.
- Collect nucleic acids by centrifugation in a microfuge at top speed at 4 °C for 20 min (see Note 28).
- Discard supernatant, spin samples briefly and remove the remaining supernatant with the pipette. Resuspend each pellet in 50 μl of ice-cold MilliQ water. At this step, all of the aliquots coming from the same original sample can be combined again in a single 1.5 ml tube that will contain 500 μl sample. Do not proceed to the next step until the pellets are completely resuspended.
- Add 500 μl of 4 M LiCl, mix by inversion and let sit on ice for 30 min or longer (see Note 29).
- Collect the RNA by centrifugation in a microfuge at 4 °C at top speed for 30 min, and discard the DNA-containing supernatant.
- Wash the pellet with 500 μl ice cold 70% ethanol, centrifuge at top speed for 10 min, discard the supernatant, and air-dry the pellets for 10-15 min.
- Dissolve RNA in 50 μl of ice-cold RNase-free water.
- Quantify RNA yield in a spectrophotometer at 260 nm and check quality in a 1% agarose TAE gel.
- Keep the total RNA samples at -80 °C until used to purify mRNA.
B2. Purification of the mRNA
Purify mRNA from total RNA using oligo-dT-coated magnetic beads from the Dynabeads mRNA Purification Kit essentially as described by the manufacturer.
- Prepare 220 μl of 1x binding buffer by diluting it from the 2x binding buffer stock provided with the Dynabeads Kit.
- Resuspend the magnetic beads by vortexing the vial and transfer 150 μl per sample to a 1.5 ml non-stick tube.
- Collect beads by placing the tube on the magnetic rack for 30 sec and carefully pipette away the storage buffer.
- Immediately resuspend beads in 100 μl of 1x binding buffer. Repeat this procedure to wash beads again in 1x binding buffer and leave them in binding buffer.
- Take 100-150 μg of total RNA and dilute to a final volume of 50 μl with RNase-free water.
- Add 50 μl of 2x binding buffer and denature the RNA for 2 min at 80 °C.
- Place the samples immediately on ice and add 1 μl of SUPERase-In.
- Remove carefully the binding buffer from the magnetic beads (step B2.4), add the entire RNA sample to the beads and resuspend carefully with the pipette.
- Incubate for 5 min at room temperature with gentle agitation in an orbital shaker to allow mRNA to bind to the beads.
- Return the tube to the magnetic rack to collect the beads for 30 sec and pipette away the unbound sample.
- Wash the beads twice with 100 μl of wash buffer B provided with the Dynabeads Kit.
- Remove all of the wash buffer B and resuspend the beads in 20 μl of 10 mM Tris-HCl, pH 7.0 mixing carefully with the pipette.
- Elute the RNA by heating the beads to 80 °C for 2 min.
- Immediately place the tube on the magnetic rack for 30 sec and transfer the eluted mRNA to a new tube.
- Save the mRNA at -80 °C if not going directly to the alkaline lysis step (step B3).
B3. Alkaline lysis
At this step the mRNA will be randomly fragmented and then, the bands with the same size as the footprints will be size-selected (C1). It is important to empirically determine the time of fragmentation, so that the sample is properly fragmented, yet the majority of the sample is not under 28 nt (see Figure 5A).
- Prepare the mRNA fragmentation reaction by mixing 20 μl of RNA (step B2.14) with 20 μl of 2x alkaline fragmentation buffer (see Recipes).
- Incubate the fragmentation reaction for 20-40 min at 95 °C. If possible, use a thermal cycle to keep temperature stable and to ensure reproducible conditions.
- Return the reaction to ice immediately.
- Pipette 560 μl of alkaline fragmentation stop/precipitation solution (see Recipes) into a non-stick microfuge tube on ice and add 40 μl of the fragmentation reaction to the stop solution. Mix by inverting the tube.
- Add 600 μl of ice-cold isopropanol and mix by inversion (see Note 30). Precipitate for 30 min at -80 °C or longer.
- Spin at 4 °C, at 20,800 x g for 30 min.
- Discard the supernatant, let the pellet air-dry for 1 min and resuspend in 10 μl 10 mM Tris-HCl, pH 8.0.
The fragmented mRNA obtained can be kept at -80 °C until gel size selection, which is performed side-by-side with the ribosome footprints samples.
- Steps: Sequencing library preparation
C1. Size selection
At this step, footprints and fragmented mRNA can be processed in parallel. As reference for size, specifically designed RNA primers of 25 and 34 nt (oNTI265 and oNTI268 [Ingolia et al., 2009]) should be run with the samples in parallel lanes and the bands delimited by these primers selected and cut out of the gel and saved.
- Pre-run a 12-well 15% TBE-Urea gel in 1x TBE (see Recipes) in a Mini-PROTEAN tetra cell polyacrylamide gel box at 200 V for 15 min.
- Prepare gel samples
a. Add 5 μl of 2x denaturing sample buffer with dye to the 10 μl samples of footprint RNA and fragmented mRNA (from steps A4.15 and B3.7, respectively).
b. Prepare the loading control by combining 0.5 μl of 10 bp ladder, 4.5 μl of water and 5 μl of 2x sample buffer
c. Prepare the size-control RNA oligos: add 1 μl of each oligo (oNTI265 and oNTI268) diluted to 10 μM, 4 μl of MilliQ water and 5 μl of 2x sample buffer per gel lane (see Note 31). - Keep the samples on ice until all of them are prepared.
- Denature samples, ladder and oligos at 80 °C for 90 sec.
- Load samples on the gel and run for 50-65 min at 200 V (see Note 32).
- Stain the gel with 1x SYBR Gold in 1x TBE by gentle shaking for 5 min.
- Visualize the gel in a UV transilluminator. In Figure 5A, a gel image shows how footprints and fragmented RNA libraries should look like.
- Excise the 25-34 nt region (demarcated by size marker oligos) from the footprinting and mRNA samples. Excise the oligos as well (combining both bands together) to use as a control for RNA recovery. It is also useful to perform a control of ligation to the polyadenylated linker with them (step C3).
- Extract RNA from gel slices
- Place the gel slice in a 1.5 ml non-stick RNase-free microfuge tube.
- Add 400 μl of RNA gel extraction buffer (GEB) (see Recipes).
- Freeze for 15-30 min at -80 °C or on dry ice.
- Place on a room temperature nutator overnight.
- Place the gel slice in a 1.5 ml non-stick RNase-free microfuge tube.
- Spin for 1 min at maximum speed in a microfuge and recover the eluate to a new tube (see Note 33).
- Precipitate RNA from gel extraction
- Take 400 μl of the eluate, add 1.5 μl of GlycoBlue, mix by inversion, and add 500 μl of isopropanol.
- Precipitate at least for 30 min at -80 °C or on dry ice.
- Spin at 4 °C at 20,800 x g for 30 min.
- Decant supernatant carefully and then spin briefly to collect the supernatant at the bottom of the tube. Remove the residual liquid with the micropipette.
- Air-dry the pellet for 1 min at room temperature.
- Take 400 μl of the eluate, add 1.5 μl of GlycoBlue, mix by inversion, and add 500 μl of isopropanol.
- Resuspend the RNA in 10 μl of 10 mM Tris-HCl, pH 8.0 and transfer to a clean microfuge tube.
C2. Dephosphorylation
This dephosphorylation step is necessary to allow ligation of the 24-35 nt RNA fragments to the Universal miRNA linker.
- Add 33 μl of ice-cold MilliQ water to the 10 μl RNA (step C1.11).
- Denature for 90 sec at 70 °C and then equilibrate at 37 °C.
- Add 5 μl of 10x polynucleotide kinase buffer, 1 μl of SUPERase-In and 1 μl of T4 polynucleotide kinase and mix well by pipetting.
- Incubate for 1 h at 37 °C and heat-inactivate for 10 min at 70 °C.
- Precipitate RNA by adding 39 μl of water, 1 μl of GlycoBlue, 10 μl of 3 M NaOAc, mix well by inverting the tube, and then add 150 μl of ice-cold isopropanol. Mix well by inverting the tube. Incubate at least 30 min at -80 °C or on dry ice.
- Pellet RNA, remove liquid, and air-dry as above (steps C1.10b-C1.10e)
- Resuspend the dephosphorylated RNA in 5 μl of 10 mM Tris-HCl, pH 8.0 and transfer it to a clean microfuge tube.
C3. Linker ligation
The ligation reaction requires a high concentration of the pre-adenylylated linker (Universal miRNA cloning linker). The pre-adenylated primer should be diluted to 205 ng/μl with 5 μl of Tris-HCl, pH 8.0.It is useful to monitor the extent of ligation, especially in preliminary experiments. For this, a ligation reaction can be set with the primers that have been recovered from the gel (step C1.8). This also gives an idea of how well gel recovery worked.
- Prepare the ligation reaction mix with excess (see Note 34). For each reaction, mix 2 μl of 10x T4 Rnl2 buffer, 1 μl of SUPERase-In, and 6 μl of PEG 8000 50% (w/v) (the PEG 8000 should be no more than a month old).
- Combine on ice the RNA sample (from step C2.7), 2 μl of Universal miRNA Linker 205 ng/μl and MilliQ water to a final volume of 10 μl.
- Denature for 2 min at 80 °C and then equilibrate to 37 °C.
- Add 9 μl of reaction mix from step C3.1 to each sample and mix well.
- Add 1 μl of T4 Rnl2(tr) to each sample and mix well.
- Incubate for a minimum of 2.5 h at room temperature (see Note 35).
Note: Can be day 5 if a 2.5 h ligation is performed instead of letting it sit overnight. Herein, day counting continues as if an overnight incubation was done.
- Stop ligation and precipitate RNA by adding 40 μl of 3 M NaOAc, pH 5.5, 1.5 of μl GlycoBlue, and 338 μl of MilliQ water. Mix well and then add 500 μl of isopropanol. Mix well by inverting the tube.
- Incubate at -80 °C, pellet, remove supernatant, and air-dry as described above (steps C1.9 and C1.10).
- Resuspend ligation products in 5 μl of 10 mM Tris-HCl, pH 8.0.
C4. Gel purification of the ligated products
This gel purification is used to separate ligation products from the pre-adenylated linker, which is only 17 nt long.
- Pre-run a 12-well 15% TBE-Urea gel in 1x TBE at 200 V for 15 min.
- Prepare gel samples
- Add 5 μl of 2x denaturing sample buffer to the 5 μlRNA samples (from step C3.9).
- Prepare the 10 bp ladder and the size control oligos (oNTI265 and oNTI268) as in steps C1.2b and C1.2c.
- Add 5 μl of 2x denaturing sample buffer to the 5 μlRNA samples (from step C3.9).
- Keep the samples on ice until all of them are prepared.
- Denature samples, ladder and oligos at 80 °C for 90 sec.
- Load samples on gel and run for 50-65 min at 200 V (see Note 36).
- Stain the gel for 5 min in 1x TBE, 1x SYBR Gold while shaking gently in an orbital shaker.
- Visualize the gel in a UV transilluminator and excise the desired bands, which will now be 17 nt longer than the input. Figure 5B shows an image of the ligated samples.
- Extract RNA from the gel slices as described above (step C1.9).
- Precipitate RNA from the gel extraction as described above (step C1.10).
- Resuspend purified ligation product in 10 μl of 10 mM Tris-HCl, pH 8.0.
C5. Reverse transcription
The reverse transcription (RT) is performed with the SuperScript III Reverse Transcriptase Kit from Invitrogen. RT primer NI-NI-9 (Ingolia et al., 2009) should be present in modest excess of target RNA (see Note 37).
- Prepare at room temperature a reaction mix containing (per reaction) 4 μl of 5x first-strand buffer, 1 μl of MilliQ water, 1 μl of dNTPs (10 mM each), 1 μl of 100 mM DTT, 1 μl of SUPERase-In and 1 μl SuperScript III. Allow some excess for the mix.
- Add 1 μl of the 1.25 μM primer NI-NI-9 to the 10 μl ligated RNA (from step C4.9).
- Denature the RNA and primer for 2 min at 80 °C, then place reactions on ice.
- Add 9 μl of the RT reaction mix and mix well.
- Incubate for 30 min at 48 °C.
C6. Removal of RNA template
- Add 2.2 μl of 1 N NaOH to each reaction. Mix well.
- Incubate at 98 °C for 20 min.
- Neutralize the reaction by adding 20 μl of 3 M NaOAc, pH 5.5, 1.5 of μl GlycoBlue, and 156 μl of MilliQ water. Mix well by inverting the tube.
- Immediately add 300 μl of isopropanol and mix well by inverting the tube.
- Precipitate and pellet cDNA, remove the supernatant, and air-dry pellets as described above for RNA (steps C1.10b-C1.10e).
- Resuspend the cDNA pellet in 5 μl of 10 mM Tris-HCl, pH 8.0.
C7. Gel purification of product
This gel purification step is necessary to separate the RT primer, which runs around at 100 nt, from the extended products, which may be as short as 128 nt. The poor separation of these bands is the reason that a large excess of primer causes background.
- Pre-run a 12-well 15% TBE-Urea gel in 1x TBE at 200 V for 15 min.
- Prepare gel samples: add 5 μl of 2x denaturing sample buffer to the 5 μl of cDNA from the RT (step C6.6). Prepare the 10 bp ladder as described before (step C1.2b). Prepare the size control by combining 2 μl of unextended 1.25 μM NI-NI-9 primer with 4.5 μl of water and 5 μl of 2x denaturing sample buffer.
- Denature samples at 98 °C for 90 sec.
- Load samples on gel and run for 50-60 min at 200 V.
- Stain the gel for 5 min in 1x TBE, 1x SYBR Gold on a gentle shaker.
- Visualize the gel and excise the extended RT product bands on a UV transilluminator. Figure 5C shows the gel image of reverse-transcribed samples and the NI-NI-9 oligo control.
- Extract DNA from the gel slices as described above (step C1.9), but use DNA gel extraction buffer (STE) (see Recipes), instead of GEB.
- Precipitate DNA from the gel extraction as described above for RNA (step C1.10).
- Resuspend reverse transcription products in 15 μl of 10 mM Tris-HCl, pH 8.0.
C8. Circularization
For the circularization step, the CircLigase ssDNA ligase from Epicentre is used.
- Prepare a reaction mix with some excess. For one reaction, use 2 μl of 10x CircLigase buffer, 1 μl of 1 mM ATP, 1 μl of 50 mM MnCl2 and 1 μl of CircLigase, and mix well.
- Add 4 μl of reaction mix to the 15 μl of resuspended cDNA samples from step C7.9 and mix well.
- Incubate for 1 h at 60 °C.
- Heat-inactivate for 10 min at 80 °C.
C9. Subtractive hybridization
The products of the circularization reaction can be used directly as a template for PCR amplification of a complete sequencing library. For ribosome footprinting samples, the circularization reaction can instead be used as a direct input to subtractive hybridization to remove high-abundance rRNA-derived sequences. A first round of footprints sequencing can be performed to analyze which are the most abundant rRNA sequences, and with that information, design biotinylated oligos to remove those abundant rRNA sequences in the future experiments.
- Prepare the Subtraction Pool with 2 μl of each distinct biotinylated rRNA subtraction oligo from the 200 μM oligo stocks (see Materials and Reagents) and add water to a final volume of 40 μl. This pool can be stored at -20 °C indefinitely.
- Combine 10 μl of circularized libraries from step C8.4 with 2 μl of the Subtraction Pool, 2 μl of 20x SSC (see Recipes) and 6 μl of MilliQ water (see Note 38).
- Denature the samples at 100 °C for 90 sec and anneal by lowering the temperature to 37 °C at a ramp of 0.1 °C/sec in a thermal cycler. Keep at 37 °C for 15 min.
- Dilute 500 μl of 2x subtraction bind/wash buffer (see Recipes) with 500 μl of water to make 1 ml of 1x bind/wash buffer.Resuspend the Dynabeads (10 mg/ml Invitrogen) by vortexing and use 25 μl per subtraction reaction.
- Collect beads by placing the tube on the magnetic rack for 30 sec and carefully pipette away the storage buffer.
- Immediately resuspend beads in one volume of 1x bind/wash buffer. Repeat this procedure to wash beads 2 more times in 1x bind/wash buffer. Pipette the 1x bind/wash buffer away.
- Resuspend the washed beads in 2x bind/wash buffer, 20 μl per subtraction reaction, and equilibrate at 37 °C.
- Add the 20 μl sample (step C9.3) to 20 μl of washed Dynabeads and incubate for 15 min at 37 °C with shaking.
- Recover the 35 μl eluate and transfer it to a new 1.5 non-sticky tube.
- Add 3 μl of GlycoBlue, 12 μl of 5 M NaCl, and 148 μl of water and mix well by pipetting gently.
- Precipitate by adding 150 μl of isopropanol and mix by inverting the tube (see Note 39).
- Precipitate DNA as described above (steps C1.10b-C1.10e).
- Resuspend in 10 μl of 10 mM Tris-HCl, pH 8.0.
C10. PCR amplification
PCR amplification does not substantially distort the relative representation of different sequences in the library until the point at which the primers are nearly exhausted and, presumably, there is competition between sequences for primer binding as well as for re-annealing of template strands as opposed to hybridization. For this reason, it is useful to set up PCR reactions with different cycle numbers to optimize the yield for each individual template without saturating the reaction. If a large quantity of library is needed, this preliminary optimization can be used to select amplification conditions for a larger reaction.
Once these conditions are set up, it is recommended to perform multiple independent amplification reactions for each library rather than a single reaction in a larger volume (e.g., set up a 100 μl PCR mixture, distribute it in 5 different PCR tubes, run the PCRs and then combine the products again in a single tube). Since PCR amplification bias appears to be random, the bias in the amplification becomes less severe when multiple independent reactions are combined, so that the resulting sequencing reads are distributed more evenly along the transcripts.For this amplification step, use the Phusion High-Fidelity DNA Polymerase Kit.
To multiplex multiple libraries in one sequencing round, perform the amplification PCR with Indexed oligos that allow the de-convolution of the reads of individual libraries based on their barcodes included in the oligos (see Materials and Reagents).
- Prepare a 100 μl PCR mixture by combining 20 μl of 5x HF buffer, 2 μl of 10 mM dNTPs, 0.5 μl of 100 μM NI-NI-2 primer (Ingolia et al., 2009), 0.5 μl of 100 μM NI-NI-3 (or an alternative indexed primer: different combinations of NI-NI-2 and indexed primer should be used for all libraries to be multiplexed together in a single sequencing lane), 5 μl of the 10 μl sample as template (step C9.14), 71 μl of nuclease-free water, and 1 μl of Phusion polymerase. At least for 1 primer pair prepare a non-template negative control reaction.
- Split the 100 μl mixture into five 200 μl strip tubes, 20 μl per tube, for each footprinting and mRNA library.
- Perform PCR using the following program: 30 sec initial denaturation at 98 °C, followed by X cycles of 10 sec denaturation at 98 °C, 10 sec annealing at 65 °C, and 5 sec extension at 72 °C. When determining the proper number of cycles for the PCR amplification, remove strip tubes at the end of the extension phase after 8, 10, 12, 14 and 16 cycles. For the oligo control, remove the non-template control at 12 cycles. Allow one of these non-template oligo controls to be run per gel with PCR products. When visualizing the PCR amplification in the gel, choose the best number of cycles to amplify the libraries that gives the best amplification but does not saturate the reaction (see Note 40).
- Once the optimal number of cycles is established, perform the 5 independent PCRs per library with the selected conditions.
C11. Gel purification of the PCR products
- Prepare samples for gel purification of the PCR products: add 3.3 μl of 6x non-denaturing loading dye (see Recipes) to each PCR reaction. In parallel, prepare one ladder sample for each gel: combine 1 μl of 10 bp ladder, 15.7 μl of water, and 3.3 μl of 6x dye.
- Prepare a 12-well 8% polyacrylamide non-denaturing gel (see Recipes) in 1x TBE and pre-run the gel at 180 V for 15 min.
- When setting up the conditions, load the five PCR amplifications of one template at different cycle numbers in a series of adjacent wells. This will require 10 wells in one gel for a pair of samples. Also load a 10 bp ladder sample and a 10-cycle oligo amplification sample on each gel. Once the optimal PCR conditions are determined, combine the 5 independent PCRs per library in a single tube and run two wells with 20 μl of the combined PCRs each (see Note 41).
- Run the gel for 40 min at 180 V.
- Stain the gel for 5 min in 1x TBE, 1x SYBR Gold on a gentle shaker.
- Visualize the gel and excise the amplified PCR product bands from the reaction that should have given a strong product band as well as still has remaining, unconsumed oligos (see Figure 5D). Avoid the faint no-insert band that will appear beneath the library band for some samples.
- Extract DNA from the gel slice as described above (step C7.7).
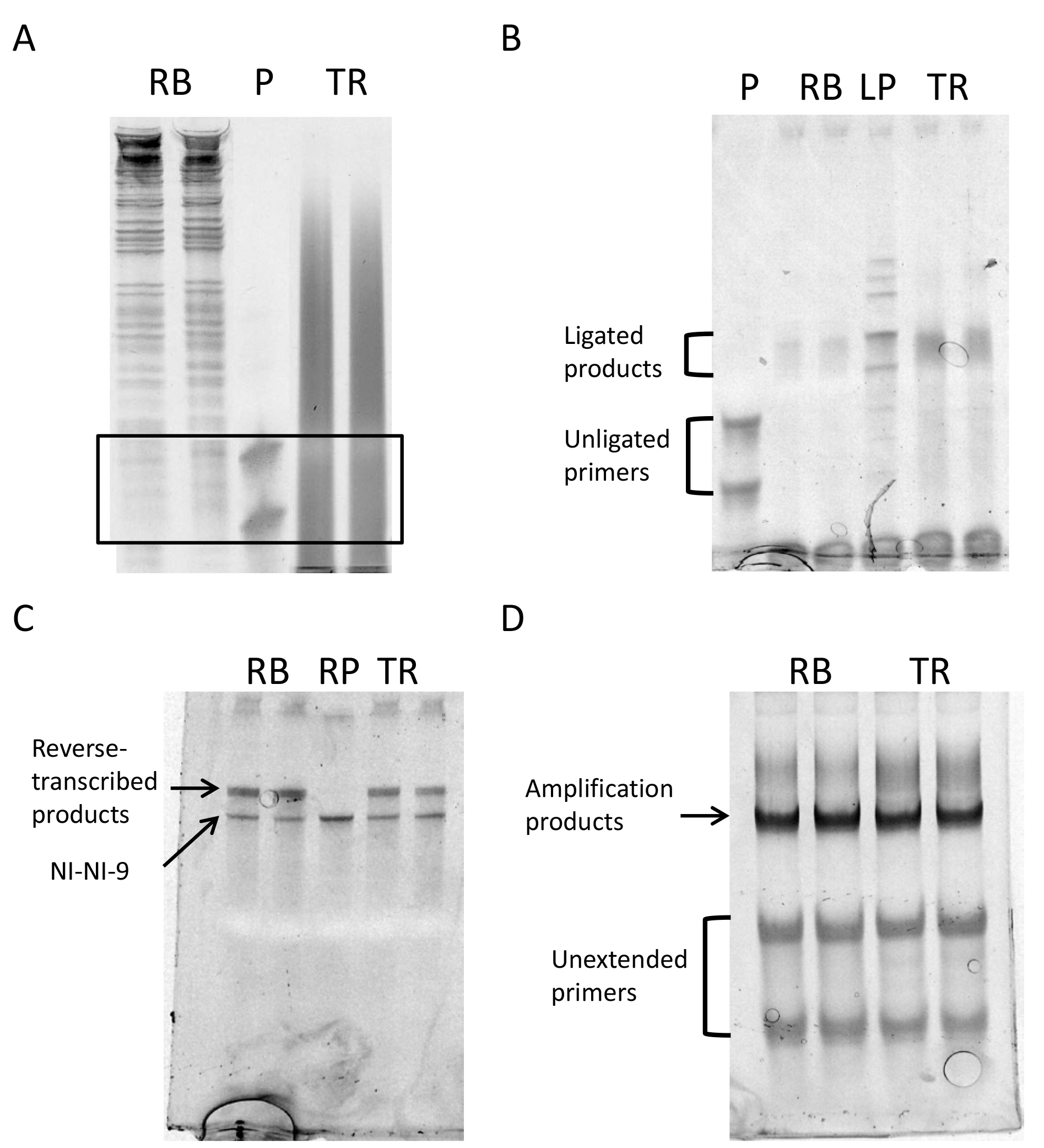
Figure 5. Gel images of different steps of the protocol. A. Size selection: The black box delimits the part of the gel to be cut out of the gel to purify the RNA; B. Gel after ligation; C. Gel after retrotranscription; D. Gel after final library amplification. RB: Ribosome footprints, P: 24-28 nt primers, TR: total mRNA fragments, LP: ligated primers as ligation control, RP: NINI-9, the primer for reverse transcription as size control.
- Precipitate, pellet, and dry the recovered DNA (steps C1.10b-C1.10e).
- Resuspend in 15 μl 10 mM Tris-HCl, pH 8.0.
C12. Sequencing library quantitation by BioAnalyzer
This step depends on the selected platform for sequencing, but the instructions presented below are mostly general.
- Check the concentration and purity of the sequencing library using the BioAnalyzer high sensitivity DNA assay.
- Use 2 μl of each library with 8 μl of water for a 1:5 dilution of each sample.
- The quantitation results are reliable only if the diluted sample concentration does not exceed 500 pg/μl. If the DNA concentration is substantially higher than this, prepare a less concentrated sample by further dilution and determine the sample concentration.
- When using pooled and barcoded libraries, adjust concentrations of the individual libraries to be pooled and then add equal amounts of each library to the desired number of HiSeq lanes. The way the libraries are multiplexed depends on the researcher’s needs. In our case, we pooled complete experiments together (2 biological replicates of mock and treatment for both footprints and total mRNA). This way, 8 barcoded libraries were combined together into 7 Illumina HiSeq lanes, with each lane run with the same mixture of 8 libraries. We typically obtain about 150 million reads per lane and between 10% to 20% of these reads correspond to useful footprints (the remaining 80% to 90% are mostly ribosomal rRNA sequences).
Data analysis
The bioinformatic analysis described herein for the study of translation regulation using ribosome footprinting data follows the pipeline described in Merchante et al. (2015). The inputs for this pipeline are the de-multiplexed Ribo-seq and RNA-seq datasets (fastq-files) from Illumina HiSeq2000 platform, the TAIR 10 Arabidopsis genome sequence, and TAIR 10 Arabidopsis gene models (https://www.arabidopsis.org/).
- General data preprocessing
- Using FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/), remove adaptor sequences and trim low-quality regions.
- Align reads to the Arabidopsis thaliana genome sequence using Tophat2 (https://ccb.jhu.edu/software/tophat/index.shtml).
- Select uniquely mapping, sense-orientation reads in the size range of 20-40 nt, discard alignments with more than two mismatches, and create read count summaries of coding (CDS) regions.
- Using FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/), remove adaptor sequences and trim low-quality regions.
- Assessment of periodicity for quality control
Ribosome footprints, but not the RNA-seq reads, shouldshow a strong 3 nt periodicity, which represents the codon-long step-wisemovement of the ribosome along the mRNA (Ingolia et al., 2009; Merchante et al.,2015). So, the presence of this periodicity can be used as a control of thequality of the footprints. To avoid edge effects, restrict analysis to thegenes with 5’ and 3’ UTRs longer than 70 nt and CDSs longer than 150 nt. Toavoid ambiguities with respect to the location of reads along a transcript,restrict analysis to reads of a fixed length - typically a value between 28 to32 nt, determined by visual inspection (C1 steps) - that map in senseorientation to only one transcript.- Transform genomic read coordinates into transcript coordinates.
- Visualize read distribution in the vicinity of the CDS start: project the selected reads into the common coordinate system of a ‘metagene’. Bin the transcript coordinates of the read start points using their relative location with respect to the start of the main open reading frame: a read that starts at the beginning of the CDS falls into bin ‘0’. To visualize the read distribution around the end of the CDS, proceed analogously but bin with respect to the end of the main open reading frame.
- Normalize the histogram of read start positions by the total number of reads selected and draw a region around bin ‘0’.
- Transform genomic read coordinates into transcript coordinates.
- edgeR analysis of differential transcription and translation
- Discard genes with small read counts (rpkm value smaller than 3 in any of the samples) and perform TMM normalization (Robinson and Oshlack, 2010).
- Model the experiment by a 2-factorial design, where factor one is, for example, ethylene/control treatment and factor two is RNA-seq/Ribo-seq treatment. Use the generalized linear models approach (glm edgeR) of the Bioconductor package edgeR (Robinson et al., 2010) to select genes that, in this example, i) respond to ethylene on the RNA level, ii) respond to ethylene on the footprint level, and iii) respond differently to ethylene on the RNA-level and the footprint level. Determine the false discovery rate using Benjamini and Hochberg’s method (Benjamini and Hochberg, 1995).
- Discard genes with small read counts (rpkm value smaller than 3 in any of the samples) and perform TMM normalization (Robinson and Oshlack, 2010).
- Analysis of changes intranslational efficiency
- Following the definition of Ingolia and colleagues (Ingolia et al., 2009), compute translational efficiency (TE) as the fraction of Ribo-seq footprint rpkm value over RNA-seq rpkm value: TE = rpkm(foot)/rpkm(mRNA).
- Compute log2-transformed TE-values of ethylene-treated plants and subtract corresponding control values: ΔTE = log2 (TEethylene) - log2 (TEcontrol)
- Compute z-score of ΔTE: zΔTE= (ΔTE-E[ΔTE])/√Var(ΔTE). Determine the false discovery rate using the R package fdrtool (Strimmer, 2008).
- Following the definition of Ingolia and colleagues (Ingolia et al., 2009), compute translational efficiency (TE) as the fraction of Ribo-seq footprint rpkm value over RNA-seq rpkm value: TE = rpkm(foot)/rpkm(mRNA).
Notes
- Cycloheximide and chloramphenicol are very toxic and harmful to the environment.
- Both phenol and chloroform are acute toxins. Handle them in a fume hood and wear proper protection.
- The 2x denaturing sample buffer with dye contains formamide, which is a reproductive toxin. Use proper protection when handling it.
- Nucleic acid stains are typically mutagenic, use proper protection when handling them.
- Both acrylamide and bisacrylamide are neurotoxins, use proper protection when handling them.
- Make sure not to confound the proper T4 PNK (M0201) with the T4 Polynucleotide Kinase 3’ phosphatase minus (M0236) as with the latter one the ligation step will not work.
- [P] represents 5’-phosphorylation and [SC18] represents a hexa-ethyleneglycol spacer.
- ‘r’ indicates that these are RNA oligonucleotides.
- These oligos may need to be re-designed if different treatments, growth conditions, developmental stages, footprinting protocols, etc., are used.
- This protocol requires Tris-HCl at pH 7.5, 7.8, 8.0, 8.4, 8.8 and 9.0, but all intermediate-concentration Tris-HCl buffers can be made out of the Tris-HCl pH7.0, 8.0 and 9.0 listed here.
- EGTA and EDTA only dissolve after adjusting the pH.
- There will be foam due to the use of detergents. Make sure to collect all the bubbles as well.
- When preparing the aliquots, allow some excess as the solution is very viscous. 1.2 ml aliquots work well for two samples.
- The SW55Ti rotor allows spinning 6 samples at the same time, so a maximum of 3 ml of SCA will be needed per centrifugation round.
- Such as a 5 ml ultracentrifuge tube or a 15 ml Falcon tube.
- This step is necessary because the debris pellet can easily get dislodged if supernatant is not transferred to a different tube rapidly at the end of the spin; failure to transfer the supernatant immediately will result in carryover of the debris and compromise ultracentrifugation.
- Sucrose gradients can also be prepared at the monosome pelleting centrifuge-step A2.7.
- Approximately half of the tube.
- The polysome pellet will be clear if using etiolated seedlings and dark green if photosynthetic tissues are employed.
- If there is a control tube that is not going to be digested, do not add the RNase I to it and keep it on ice until digestion time is over.
- Be extra-careful when doing this to not disturb the gradient.
- If using 1.5 ml tubes to heat the phenol:chloroform, do not allow the temperature to get higher than 65 °C and secure the lids, as, due to the pressure, the lids can open quite easily and produce dangerous spills, leading to the loss of precious samples.
- Phenol alone can also be used to extract the RNA, but if this is the case, the aqueous phase can be the lower one due to the high concentration of sucrose in the extract.
- Other standard RNA extraction methods (e.g., Trizol) compatible with high-throughput sequencing library construction and next-gen sequencing can also be used.
- Phenol:Chloroform:Isoamylalcohol (25:24:1) can also be used.
- JA17 tubes in the Beckman Avanti J-25 can also be used.
- If using 2 ml centrifuge tubes, add 500 μl of extract to different microtubes prefilled with 50 μl 3 M sodium acetate pH 5.2, mix and add 1,375 μl of ethanol.
- 34,895 x g if the Beckman centrifuge is used.
- At this step, samples can be left overnight at 4 °C if necessary.
- The stop solution already contains 300 mM salt, so no more salt is needed to precipitate the RNA.
- It is easier to select the size if the samples are surrounded by the oligos, so, run two oligo lanes, at the beginning and end of the gel.
- The time of the run has to be determined empirically depending on the running cell. It is a good practice to start running a test with the primers. As a reference, for the first run do not let the lower blue band from the denaturing sample buffer run out of the gel. Note the position of the marker primers with respect to the dye and adjust the future run times to the primer test.
- The gel piece does not disintegrate or change appearance.
- Allow a relatively high excess as the mix is very viscous and difficult to pipette.
- If the room-temperature incubation is followed by an overnight incubation at 4 °C, ligation efficiency increases.
- As a reference, the ligated samples typically run just above the higher blue band of the sample buffer.
- When the primer is present in vast excess, there is increased background from unextended primer that can, in some cases, dominate the actual library products. It is best to determine the right proportion empirically. 1-2 μl of NI-NI-9 primer at 1.25 μM should work fine. This can be tested in the gel run afterwards: the unextended primer should still be present after the RT step, but the extended libraries should not be fainter than the primer itself.
- The circularized libraries were resuspended in 20 µl and only 10 µl are used for the subtractive hybridization step. Keep the remaining 10 µl at -80 °C as a back-up.
- The buffer conditions in the subtractive hybridization (high salt as well as citrate, which chelates magnesium) are incompatible with PCR and thus precipitation is required after the subtraction step.
- Look for the cycle number that results in the best amplification without a significant loss of primers, so that there won’t be competition between sequences for the primers. The conditions are usually stable between experiments. Therefore, once the optimal conditions have been determined for a subset of samples, they also prove suitable for other samples from different experiments. In our case, the best amplification was achieved at 14 cycles, and further tests with additional samples confirmed 14 cycles as optimal, negating the need for further optimization.
- Keep the rest of the PCR as a back-up.
Recipes
- Polysome extraction buffer (PEB)
110 mM Tris-HCl, pH 8.8
100 mM sucrose
100 mM KCl
75 mM NaCl
20 mM MgCl2
12.5 mM EGTA
3 mM DTT
6.25 μl/ml detergent mix
25 μl/ml Triton X-100
37.5 μg/ml cycloheximide
25 μg/ml chloramphenicol
Prepare fresh before the extraction and keep on ice until use - Sucrose cushion A (SCA)
1.75 M sucrose
400 mM Tris-HCl, pH 9.0
200 mM KCl
5 mM EGTA
35 mM MgCl2
5 mM DTT
50 μg/ml cycloheximide
50 μg/ml chloramphenicol
Prepare in advance (minus DTT and antibiotics). Autoclave no longer than 15 min, make 1.2 ml aliquots and store at -20 °C. Add the last three compounds fresh for each experiment before use. - Sucrose gradients solutions
50%/10% (w/v) sucrose
40 mM Tris-HCl, pH 8.4
20 mM KCl
10 mM MgCl2
50 μg/ml cycloheximide
50 μg/ml chloramphenicol
1 μg/ml SUPERase-In
Prepare fresh on the day of the experiment - Polysome digestion buffer (PDB)
20 mM Tris-HCl, pH 7.8
150 mM NaCl
5 mM MgCl2
50 μg/ml Triton X-100
1 mM DTT
100 μg/ml cycloheximide
12 μl TURBO DNase 2 U/μl per 4.5 ml of digestion volume
10 μl RNase I per 4.5 ml of digestion buffer
Prepare on the day of the extraction and keep on ice - Sucrose cushion B (SCB)
1.75 M sucrose
20 mM Tris-HCl, pH 7.8
150 mM NaCl
5 mM MgCl2
5 mM DTT
50 μg/ml cycloheximide
50 μg/ml chloramphenicol
1 μl/ml SUPERase-In
Prepare in advance (minus the last four ingredients) and autoclave no longer than 15 min. Make 1.2 ml aliquots and store at -20 °C. Add the last four compounds fresh for each experiment before use. - Polysome resuspension buffer (PRB)
200 mM Tris-HCl, pH 8.4
200 mM KCl
25 mM EGTA
35 mM MgCl2
50 μg/ml cycloheximide
50 μg/ml chloramphenicol
1 μl/ml SUPERase-In
Prepare the day of the extraction and keep in ice - Total RNA extraction buffer (TREB)
200 mM Tris-HCl, pH 8.0
400 mM LiCl
25 mM EDTA
1% SDS
Prepare in advance (the solution keeps for several months) and store at room temperature - Alkaline fragmentation buffer (2x)
2 mM EDTA
100 mM Na2CO3, pH 9.2
This solution is prepared by mixing 15 parts of 100 mM Na2CO3 with 110 parts of 100 mM NaHCO3. It will equilibrate with gaseous CO2 and raise in pH over time. It can be stored in tightly capped, single-use aliquots at room temperature for over a year. - Alkaline fragmentation stop/precipitation solution
60 μl 3 M NaOAc
2 μl GlycoBlue 15 mg/ml
500 μl RNase-free water per reaction
Prepare right before use - TAE (Tris/acetate/EDTA buffer) (50x)
2 M Tris base
1 M acetic acid
50 mM EDTA - TBE (Tris/Borate/EDTA buffer) (5x)
445 mM Tris base
445 mM boric acid
10 mM EDTA
Dilute the concentrated stock buffer to 1x right before use. - RNA gel extraction buffer (GEB)
300 mM NaOAc, pH 5.5
1 mM EDTA
0.25% SDS - DNA gel extraction buffer (STE)
300 mM NaCl
10 mM Tris-HCl, pH 8.0
1 mM EDTA - SSC (20x)
3 M NaCl
300 mM trisodium citrate (Na3C6H5O7)
Adjusted to pH 7.0 with HCl - Subtraction bind/wash buffer (2x)
400 μl 5 M NaCl
2 μl 0.5 M EDTA
10 μl 1 M Tris-HCl, pH 7.5
590 μl water - 8% non-denaturing polyacrylamide gel (12 ml)
6.3 ml MilliQ water
3.2 ml 30% acrylamide/bisacrylamide
2.4 ml of 5x TBE
84 μl of 10% APS
10 μl TEMED - Non-denaturing loading dye (6x)
60% (v/v) glycerol
0.03% (w/v) bromophenol blue
Acknowledgments
This work was supported by NSF grants MCB 1158181 and 0519869 to J.M.A.; MCB 0923727 to J.M.A. and A.N.S.; IOS 1444561 to J.M.A., A.N.S., and S.H.; NCSU-RISF to S.H. and J.M.A.; and a Marie Curie COFUND U-Mobility postdoctoral fellowship to C.M. (co-funded by the University of Malaga and the EU 7FP GA N_246550). We are grateful to Nick Ingolia for his help with implementing and troubleshooting this protocol in plants.
References
- Branco-Price, C., Kaiser, K. A., Jang, C. J., Larive, C. K. and Bailey-Serres, J. (2008). Selective mRNA translation coordinates energetic and metabolic adjustments to cellular oxygen deprivation and reoxygenation in Arabidopsis thaliana. Plant J 56(5): 743-755.
- Benjamini, Y. and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. B 57: 125-133.
- Ingolia, N. T., Ghaemmaghami, S., Newman, J. R. and Weissman, J. S. (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324(5924): 218-223.
- Ingolia, N. T., Brar, G. A., Rouskin, S., McGeachy, A. M. and Weissman, J. S. (2013). Genome-wide annotation and quantitation of translation by ribosome profiling. Curr Protoc Mol Biol Chapter 4: Unit 4 18.
- Juntawong, P., Girke, T., Bazin, J. and Bailey-Serres, J. (2014). Translational dynamics revealed by genome-wide profiling of ribosome footprints in Arabidopsis. Proc Natl Acad Sci U S A 111(1): E203-212.
- Li, R., Sun, R., Hicks, G. R. and Raikhel, N. V. (2015). Arabidopsis ribosomal proteins control vacuole trafficking and developmental programs through the regulation of lipid metabolism. Proc Natl Acad Sci U S A 112(1): E89-98.
- Mustroph, A., Juntawong, P. and Bailey-Serres, J. (2009). Isolation of plant polysomal mRNA by differential centrifugation and ribosome immunopurification methods. Methods Mol Biol 553: 109-126.
- Merchante, C., Brumos, J., Yun, J., Hu, Q., Spencer, K. R., Enriquez, P., Binder, B. M., Heber, S., Stepanova, A. N. and Alonso, J. M. (2015). Gene-specific translation regulation mediated by the hormone-signaling molecule EIN2. Cell 163(3): 684-697.
- Missra, A. and von Arnim, A. G. (2014). Analysis of mRNA translation states in Arabidopsis over the diurnal cycle by polysome microarray. Methods Mol Biol 1158: 157-174.
- Reynoso, M. A., Juntawong, P., Lancia, M., Blanco, F. A., Bailey-Serres, J. and Zanetti, M. E. (2015). Translating Ribosome Affinity Purification (TRAP) followed by RNA sequencing technology (TRAP-SEQ) for quantitative assessment of plant translatomes. Methods Mol Biol 1284: 185-207.
- Reuber, T. L. and Ausubel, F. M. (1996). Isolation of Arabidopsis genes that differentiate between resistance responses mediated by the RPS2 and RPM1 disease resistance genes. Plant Cell 8(2): 241-249.
- Robinson, M. D. and Oshlack, A. (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11(3): R25.
- Robinson, M., McCarthy, D., and Smyth, G. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1):139-40.
- Strimmer, K. (2008). A unified approach to false discovery rate estimation. BMC Bioinformatics 9: 303.
- Zanetti, M. E., Chang, I. F., Gong, F., Galbraith, D. W. and Bailey-Serres, J. (2005). Immunopurification of polyribosomal complexes of Arabidopsis for global analysis of gene expression. Plant Physiol 138(2): 624-635.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Merchante, C., Hu, Q., Heber, S., Alonso, J. and Stepanova, A. N. (2016). A Ribosome Footprinting Protocol for Plants. Bio-protocol 6(21): e1985. DOI: 10.21769/BioProtoc.1985.
Category
Plant Science > Plant cell biology > Organelle isolation
Plant Science > Plant cell biology > Cell isolation
Cell Biology > Organelle isolation > Polyribosome
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.