- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Establishment of a Fusarium graminearum Infection Model in Arabidopsis thaliana Leaves and Floral Tissues



Published: Vol 6, Iss 14, Jul 20, 2016 DOI: 10.21769/BioProtoc.1877 Views: 13823
Reviewed by: Arsalan DaudiSara Posé Shweta Kalve
Original research article
The authors used this protocol in:

Oct 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Fusarium graminearum (Fg) is the causal agent of Fusarium head blight disease of wheat (Triticum aestivum), oats (Avena sativa) and barley (Hordeum vulgare), which targets the floral tissues and thereby adversely impacts grain yield and quality. Mycotoxins produced by F. graminearum further limit the consumability of infected grain. In the laboratory, F. graminearum also has the ability to colonize both leaves and inflorescence tissues of Arabidopsis thaliana. The interaction between A. thaliana and F. graminearum makes available a large array of genetic and molecular tools to study the interaction between plants and F. graminearum to elucidate plant genes and pathways that contribute to resistance, as well as study how the fungus targets plant genes and mechanisms to promote disease. The methods described below allow for efficient infection of Arabidopsis leaves and inflorescence, and evaluation of disease progress and fungal growth. Disease spread in Arabidopsis can be readily monitored by the visual observations of chlorosis of leaf tissue and disease phenotype of inflorescence tissue including fungal mass on surface of the inflorescence tissue. Fungal growth can be further monitored by measuring the relative amount of Fg DNA in the host tissue by polymerase chain reaction (PCR) and quantitative real-time PCR (qPCR).
Materials and Reagents
- PCR tubes (Fisher Scientific, catalog number: 14222 262 )
- Petri dishes (100 x 15 mm) (Fisher Scientific, catalog number: FB0875713 )
- 50 ml plastic screw-capped tubes (Midsci, catalog number: C50B )
- Pipette tips (sterile) (Midsci, catalog number: AVR-1, AVR-4 and AVR-11 )
- 1.7 ml microfuge tubes (sterile) (Catalog number: AVSS1700 )
- Cheesecloth from a local craft store or Miracloth (EMD Millipore, catalog number: 475855-1R )
- Culture tube
- 1 ml needle-less syringe (Tuberculin syringe) (Becton Dickinson, catalog number: 309659 )
- Funnel
- 1 L glass conical flask (Pyrex brand)
- Tweezers
- Hemocytometer
- Camel hair brush
- Sharpie or comparable water-proof marker
- Disposable gloves
- Kimwipes, tissue paper or paper towels
- Face shield (Fisher Scientific, catalog number: 18-999-4542 )
- Kord brand 3.5 inch square pots with bottom holes (Hummert International, catalog number: 12-1350-1 )
- T.O. Plastics Standard Flats 1020 tray with bottom holes (Hummert International, catalog number: 11-3000-1 )
- T.O. Plastics Standard Flats 1020 tray without holes (Hummert International, catalog number: 11-3050-1 )
- DOM1020 plastic dome to fit 1020 flats (Hummert International, catalog number: 11-3360-1 )
- Transparent plastic bags (Glad 13 gallon Recycling Drawstring Clear Trash bag)
- Fusarium graminearum isolate Z-3639 (Bowden and Leslie, 1999)
- Arabidopsis thaliana seeds (Accession Columbia, Nössen, and Wassilewskija)
- Silwet L-77 (Lehle seeds, catalog number: VIS-30 )
- Potato Dextrose Broth (Becton Dickinson, catalog number: 254920 )
- Yeast extract (Becton Dickinson, catalog number: 212750 )
- BD Difco Agar (Becton Dickinson, catalog number: 214530 )
- Ammonium Nitrate (Fisher Scientific, catalog number: A676 )
- Potassium chloride (Fisher Scientific, catalog number: P217 )
- Magnesium sulfate heptahydrate (Fisher Scientific, catalog number: M63 )
- Sodium chloride (Fisher Scientific, catalog number: BP358-1 )
- Tris-Base (Fisher Scientific, catalog number: BP152 )
- Ethylenediaminetetraacetic acid, disodium salt, Dihydrate (Fisher Scientific, catalog number: S311 )
- Sodium dodecyl sulfate (Fisher Scientific, catalog number: BP166 )
- Carboxymethyl cellulose, CMC (Sigma-Aldrich, catalog number: C5678 )
- Sterile deionized water (dH2O)
- Sterile double distilled water (ddH2O)
- Phenol (Fisher Scientific, catalog number: BP226500 )
- Chloroform (Fisher Scientific, catalog number: C607-4 )
- Isopropanol (Fisher Scientific, catalog number: A451SK-4 )
- Ethanol (Fisher Scientific, catalog number: A995-4 )
- Primers (Listed below in Table 1)
- dNTPs (Sigma-Aldrich, catalog number: DNTP100A-1KT )
- Polymerase for PCR (Fisher Scientific, catalog number: FB-6000-10 )
- iTaq Univeral SYBR Green Supermix (Bio-Rad, catalog number: 1725122 )
- Agarose (Fisher Scientific, catalog number: BP1356 )
- Soil mix (Fafard, catalog number: Fafard Growing Mix 2/C-2 )
- Peters 20:20:20 General Purpose fertilizer (Hummert International; catalog number: 07-5400-1 )
- F. graminearum macroconidia suspension (see Procedure)
- F. graminearum mycelial fragments (see Procedure)
- Potato Dextrose Agar-Half strength (½ PDA) (see Recipes)
- Carboxymethyl Cellulose (CMC) media (see Recipes)
- Arabidopsis DNA extraction buffer (see Recipes)
- Tris-equilibrated phenol-chloroform (see Recipes)
- Spray spore suspension (see Recipes)
Equipment
- Hand-held atomizer
- Micropipettes (P20, P100 and P1000)
- Standard Lab Incubator for cultivating fungus (Fisher Scientific, Fisher ScientificTM IsotempTM)
- Plant growth chamber for cultivating Arabidopsis (Percival scientific, model: AR-66L2 )
- Thermal cycler (Techne, model: 3PrimeX )
- Real-time PCR system (Illumina, EcoTM, catalog number: EC-101-1001 )
Note: This product has been discontinued by the manufacturer. - Compound microscope (Leica, model: DM2000 )
- Tabletop centrifuge (Beckman)
- Microfuge (Fisher Scientific, Fisher ScientificTM accuSpinTM, model: Micro 17/Micro 17R )
- Vortex-Genie 2 (Scientific Industries, catalog number: SI-0236 )
- Basic power supply gel electrophoresis powerpack, trays and combs, (Bio-Rad, PowerPacTM, catalog number: 1645050 )
- Gel electrophoresis system (Bio-Rad, catalog number: 1704405 )
Software
- Variance (ANOVA) (P < 0.05) (SAS Institute Inc, SAS v5.1)
Procedure
Experiments in our lab have shown that placing F. graminearum mycelia or macroconidia on the leaf surface does not yield uniform infection. Further, the level of infection is also highly variable. However, leaves when infiltrated with fungal mycelium fragments, which are small enough to enter the leaf tissue presumably through stomatal openings, resulted in reproducible infection. Macroconidia on the other hand are larger and could not be easily infiltrated into the leaves. On Arabidopsis floral tissue, macroconidia are able to germinate and successfully infect tissue.
- Cultivation of Fusarium graminearum isolate Z-3639 and preparation of fungal mycelial suspension for inoculation of Arabidopsis leaves
- The fungus is cultivated and maintained on ½ strength Potato Dextrose Agar (PDA) made with 0.7% Agar. Plates are 100 mm (wide) x 15 mm (deep).
- To prepare fungi for inoculation, culture the F. graminearum isolate Z-3639 (Bowden and Leslie, 1999) on ½ PDA plates for 8-10 days at 28 °C. As the fungal mass grows, it turns a pinkish red color (Figure 1).
Notes:- If a 28 °C incubator is not available the fungus can be cultivated at room temperature, although preferably not below 22 °C.
- Plates with fungal mass that are older than 1 month should not be used in the preparation of inoculum.
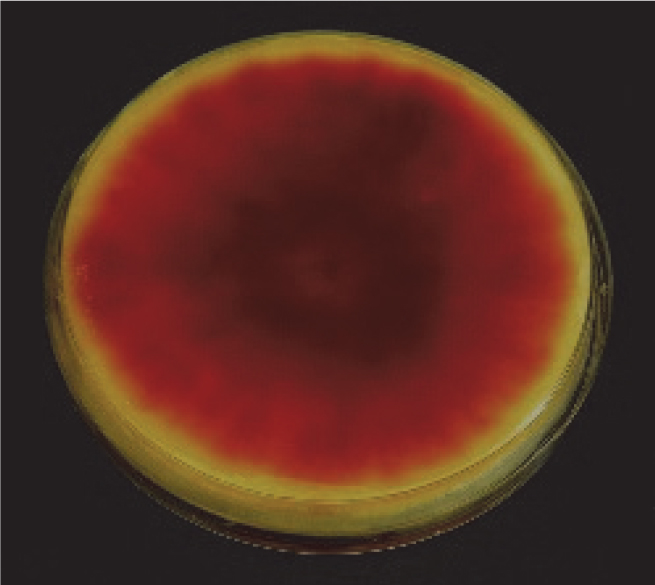
Figure 1. PDA plates showing red coloration due to Fusarium graminearum growth for 8 days at 28 °C - After 10 days, flood each plate with 10 ml of sterile ddH2O, and carefully scrape mycelia from plate surface with a soft camel hair brush taking care not to scrape off the media (see Figure 2). This process harvests fungal mycelia from the media and simultaneously fragments it into smaller pieces, which is critical for the subsequent infection of Arabidopsis leaves. The fungal suspension will have a pink color to it (see Figure 2).
Note: For control inoculations, use ½ PDA plates that were not inoculated with F. graminearum. - The fungal suspension is filtered through four layers of cheesecloth (alternatively can use two layers of miracloth) to remove debris and larger mycelial mass (see Figure 2). Finally, after suspensions from 6-7 plates are collected, pass 5 ml of sterile ddH2O through the cheesecloth (or miracloth). Repeat this wash an additional time. Typically around 50 ml of suspension is required to infiltrate 60-70 leaves.
Figure 2. Preparation of fungal mycelial fragments from ½ PDA plates. Left panel: Harvesting fungal mycelia from ½ PDA plates with a camel hair brush. Middle panel: Filtering fungal mycelial suspension through cheesecloth. Right panel: Culture tube with filtered fungal mycelial suspension.
- The fungus is cultivated and maintained on ½ strength Potato Dextrose Agar (PDA) made with 0.7% Agar. Plates are 100 mm (wide) x 15 mm (deep).
- Cultivation of Fusarium graminearum isolate Z-3639 and preparation of fungal spores for inoculation of Arabidopsis floral tissues
- Cultivate F. graminearum on ½ PDA plates for 8-10 days at 28 °C as described above.
- To promote sporulation, a 1/4th square inch of fungal plug of fungal mycelial mass is cut from the PDA plate that shows profuse fungal growth and placed in a 1 L conical flask containing 250 ml of sterile carboxymethyl cellulose media.
- Incubate the fungus-inoculated CMC media (see Recipes) containing flask on a shaker at 100 rpm at 28 °C for 7-9 days till profuse macroconidiation is observed.
- The fungal suspension is filtered through four layers of cheesecloth to remove debris and mycelial mass.
- The filtrate containing macroconidia is centrifuged in a table top swing-bucket centrifuge at 3,000 x g for 10 min.
- The pelleted macroconidia are washed by re-suspending them in 10 ml sterile ddH2O followed by centrifugation at 3,000 x g for 10 min at room temperature, as described above. This wash is repeated one more time.
- The pelleted macroconidia (Figure 3) is re-suspended to a concentration of 1 x 105 macroconidia/ml in sterile ddH2O water containing 0.001% Silwet L-77.

Figure 3. Fusarium graminearum macroconidia. Bar represents 20 μm.
- Cultivate F. graminearum on ½ PDA plates for 8-10 days at 28 °C as described above.
- Arabidopsis cultivation
- A compost-peat-based Fafard #2 soil mix was used for cultivating Arabidopsis. The soil was first sterilized by autoclaving as follows: Soil sufficient to half-fill an autoclave bag is mixed with sufficient water to until complete saturation. At the same time, care must be taken to break large clumps of soil to ensure uniform soil saturation.
- The loosely closed bag is autoclaved for 1 h. The soil is then allowed to cool to room temperature (overnight) before use.
- The autoclaved, but cooled soil was loosely packed into Kord brand 3.5” square pots with bottom holes that were placed in 20-9/16” x 10-3/16” x 2-3/8” 1020 tray with bottom holes, which were further placed in 20-9/16” x 10-3/16” x 2-3/8” 1020 tray without holes (see Figure 4).
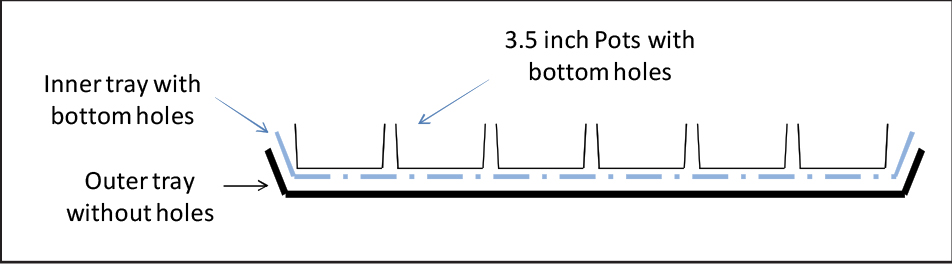
Figure 4. Arabidopsis cultivation set-up consisting of 3.5” pots contained in a tray with holes, which in turn is contained in a tray without holes - The soil was sub-irrigated by filling the outermost tray with tap water containing at 0.4 g/gallon of Peters 20:20:20 fertilizer and placing the soil-filled pots contained in the tray with bottom holes in it.
- The soil was allowed to wet by capillary action till the soil surface was well wetted.
- Excess water was drained by lifting the tray with bottom holes containing the pots above the water level.
- The water in the flat without holes was drained off.
- The drained pots in the flat with bottom holes were returned to the tray without holes.
- Two seeds per pot were placed on the surface of the soil with a moistened toothpick, one seed at a time.
- After all pots were seeded, the entire set up of pots in trays was covered with a transparent DOM1020 plastic dome and transferred into a cold room where they were left in the dark for stratification.
- Two days later, the trays with the pots were moved into a growth room or growth chamber set at 22 °C under a 14 h light (80 μE m-2 sec-1)/10 h dark regime with approximately 60% relative humidity (RH).
- Approximately four-week-old plants were used for inoculating leaf tissue with fungal mycelial fragments, while 6-7 week old plants with unbranched bolts were used for inoculating the inflorescence tissue with fungal macroconidia.
- A compost-peat-based Fafard #2 soil mix was used for cultivating Arabidopsis. The soil was first sterilized by autoclaving as follows: Soil sufficient to half-fill an autoclave bag is mixed with sufficient water to until complete saturation. At the same time, care must be taken to break large clumps of soil to ensure uniform soil saturation.
- Arabidopsis leaf infection with Fusarium graminearum
- Inoculation of Arabidopsis leaves with Fusarium graminearum mycelial fragments
- Approximately four-week-old Arabidopsis plants were used. It is important to include the appropriate control genotypes in each experiment. Plants are watered the day before inoculation. Infection is typically done in the afternoon hours.
- Expanded leaves for inoculation are marked with a water-proof marker. Approximately 4-5 leaves per plant are inoculated. A minimum of 60 leaves from 15 plants of each genotype are required for each treatment (mock v/s fungus).
- A 1 ml needle-less syringe is used for infiltrating a suspension of fungal mycelial fragments into the abaxial side (underside) of the Arabidopsis leaves (Figure 5). Leaves are infiltrated on each side of the mid-vein till the entire leaf area is infiltrated. Control (mock) treatment involves water that was passed over the PDA plates without the fungus and processed similarly to the processing of the fungal culture.
Note: Use gloves, eye protection and lab coats when carrying out fungal inoculations. All waste coming in contact with the fungal culture is collected and autoclaved before disposing. - After infiltration, plants are covered with a transparent dome for 48 h to maintain high humidity and promote fungal infection.

Figure 5. Fungal infiltration into Arabidopsis leaves. Shown is fungal culture being infiltrated with a needle-less syringe into the abaxial surface (undersurface) of an Arabidopsis leaf. - Approximately four-week-old Arabidopsis plants were used. It is important to include the appropriate control genotypes in each experiment. Plants are watered the day before inoculation. Infection is typically done in the afternoon hours.
- Scoring the severity of Fusarium graminearum disease on Arabidopsis leaves
- Disease spread is seen in the leaves as a spread of chlorosis and severity is recorded 5 days post inoculation. However, since disease progression depends on the quality of the fungal inoculum, if disease progression is slow then disease severity can be monitored at day 6 or even day 7.
- The percentage of inoculated leaves exhibiting chlorosis covering < 25% (category I), 25-50% (category II), 50-75% (category III) and > 75% (category IV) of leaf area are determined for each genotype (Figure 6). PCR analysis for fungal DNA relative to plant DNA is used to confirm fungal growth over the course of infection (Figure 7 left panel) and determine correlation between disease severity and fungal growth in the diseased leaves (Figure 7 right panel).
- A minimum of 60 leaves from 15 plants of each genotype are evaluated for disease severity.
- Disease severity index is calculated using the formula
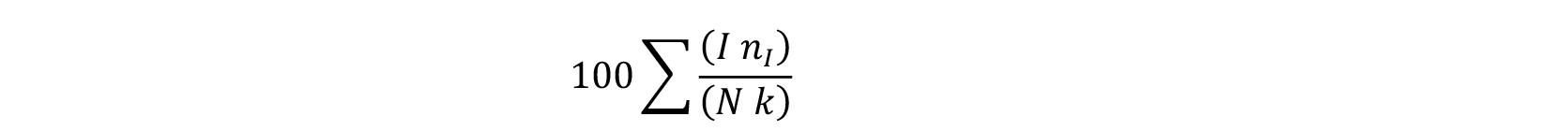
Where,
I = disease severity score: 1 for category I, 2 for category II, 3 for category III and 4 for category IV,
nI = number of leaves with each score,
N = total number of leaves,
k = highest score (in this case it is 4). - Single factor analysis of variance (ANOVA) (P < 0.05) (SAS v5.1) is used to compare disease severity amongst different genotypes. Figure 8 shows a representative data set comparing the disease severity between wild type (WT) plants of Arabidopsis accession Columbia (Col-0) and two mutants, mut-1 and mut-2, that exhibit enhanced resistance.
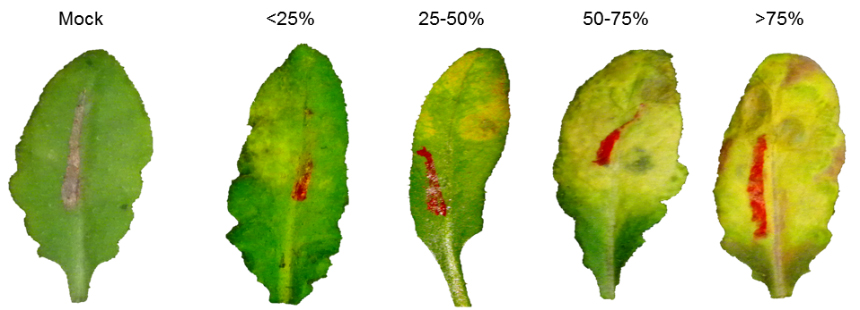
Figure 6. Disease symptoms in Fusarium graminearum-infected leaves. Diseased leaves are categorized into four groups based on the extent of leaf area exhibiting chlorosis.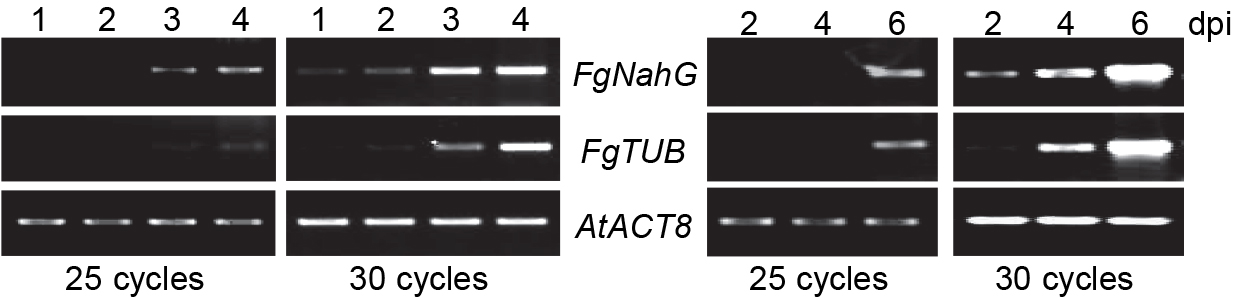
Figure 7. PCR analysis for fungal DNA. Left panel: PCR (25 and 30 cycles) for fungal and plant genes on DNA extracted from infected leaves exhibiting < 25% (lane 1), 25-50% (lane 2), 50-75% (lane 3), and > 75% (lane 4) chlorosis. Right panel: PCR (25 and 30 cycles) for fungal and plant genes on DNA extracted from 15 pooled leaves collected at 2, 4 and 6 days post inoculation (dpi). PCR was conducted with primers specific for the fungal NahG (FgNahG) and TUBULIN (FgTUB) genes, and as a control the Arabidopsis ACT8 (AtACT8) gene (served as a control).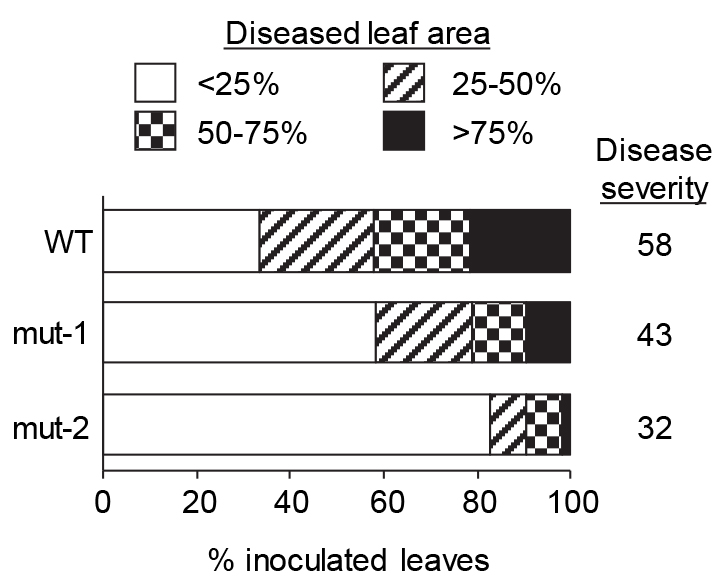
Figure 8. Fusarium graminearum disease severity in leaves of Arabidopsis wild-type accession Col-0 and two mutants (mut-1 and mut-2) that exhibit reduced disease severity. The percentage of inoculated leaves exhibiting chlorosis covering < 25%, 25-50%, 50-75% and > 75% of the leaf area at 5 dpi was determined for each genotype. The disease severity index for each genotype is indicated on the right. - Disease spread is seen in the leaves as a spread of chlorosis and severity is recorded 5 days post inoculation. However, since disease progression depends on the quality of the fungal inoculum, if disease progression is slow then disease severity can be monitored at day 6 or even day 7.
- Monitoring fungal growth by PCR
Infected leaf tissue is processed to extract DNA, which includes both, plant and fungal DNA.- DNA extraction
- Fifteen fungus-inoculated and as control mock-inoculated leaves per Arabidopsis genotype are randomly harvested, pooled and quick frozen in liquid nitrogen.
- The frozen tissue is ground to a powder in a chilled mortar with a chilled pestle. Approximately 50 mg of the frozen powder is transferred into a 1.7 ml microfuge tube containing 200 μl of DNA extraction buffer (see Recipes) at room temperature and mixed and left at room temperature for a minimum of 5 min to allow tissue dissociation.
- Once all samples are processed, they are placed in a microfuge and centrifuged at 16,500 x g for 5 min to pellet cell debris.
- The supernatant is transferred into a fresh 1.7 ml microfuge tube and mixed with 100 μl of Tris-equilibrated phenol-chloroform (pH 7-8) (see Recipes). Samples are centrifuged at 16,500 x g for 10 min at room temperature in a microfuge.
- The DNA containing supernatant is transferred into a fresh tube containing 150 μl of isopropanol. Vortex for 5 sec and leave samples at room temperature for 10 min.
- Pellet DNA by centrifugation at 16,500 x g for 10 min at room temperature. Wash pellet with 500 μl 70% ethanol and then let pellet air dry left inverted on Kimwipes for 10 min.
- Dissolve the pellet, which contains a mix of plant and fungal DNA in 200 μl of ddH2O.
- PCR for monitoring fungal growth in Arabidopsis leaves
- Primers designed to the fungal genes FgNahG and FgTUB are used to monitor the amount of fungus relative to Arabidopsis ACT8.
- 1 μl of DNA extracted from plant tissue is used for PCR in a total 20 μl volume containing 0.25 μM each of dATP, dTTP, dCTP and dGTP, 0.05 μM of each primer, and 1 unit of Taq Pol (or related polymerase) along with the appropriate PCR buffer.
- PCR was conducted using the following amplification protocol: 3 min at 94 °C for denaturation of nucleic acids, followed by 25 or 30 cycles of 94 °C for 30 sec, 58 °C for 30 sec and 72 °C for 30 sec, culminating with a step at 72 °C for 30 sec and a hold step at 4 °C.
- The PCR products were resolved on 1.5% agarose gel, stained with ethidium bromide and visualized under UV illumination (see Figure 7 as an example).
- qPCR-based quantification of fungal growth in Arabidopsis leavesQuantitative PCR (qPCR) for fungal genes (FgNahG and FgTUB) was performed with Sybr® Green PCR Master Mix on an Eco Illumina system (or any comparable real-time PCR machine) using the following amplification protocol: 10 min at 95 °C for polymerase activation and denaturation of nucleic acids, followed by 40 cycles of 95 °C for 10 sec, 58 °C for 30 sec and 72 °C for 30 sec. This was followed by a product melt to confirm a single PCR product. The level of gene expression was normalized to that of Arabidopsis EF1α by subtracting the CT value of EF1α from the CT value for the fungal gene. The ΔΔCT method (Livak and Schmittgen, 2001) was used to calculate relative fold changes. See Figure 9 as an example.
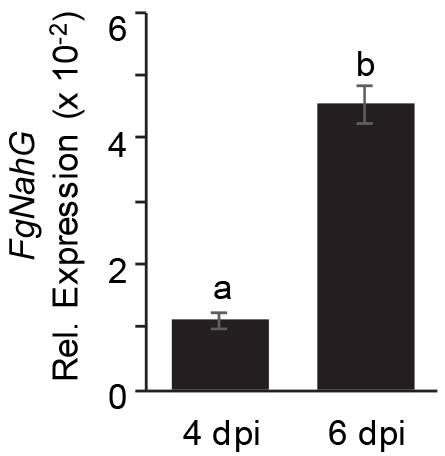
Figure 9. qPCR analysis to monitor fungal growth. qPCR analysis of fungal FgNahG gene relative to that of Arabidopsis EF1α conducted on DNA extracted from Fg-infected leaves at 4 and 6 dpi.
Table 1. PCR primers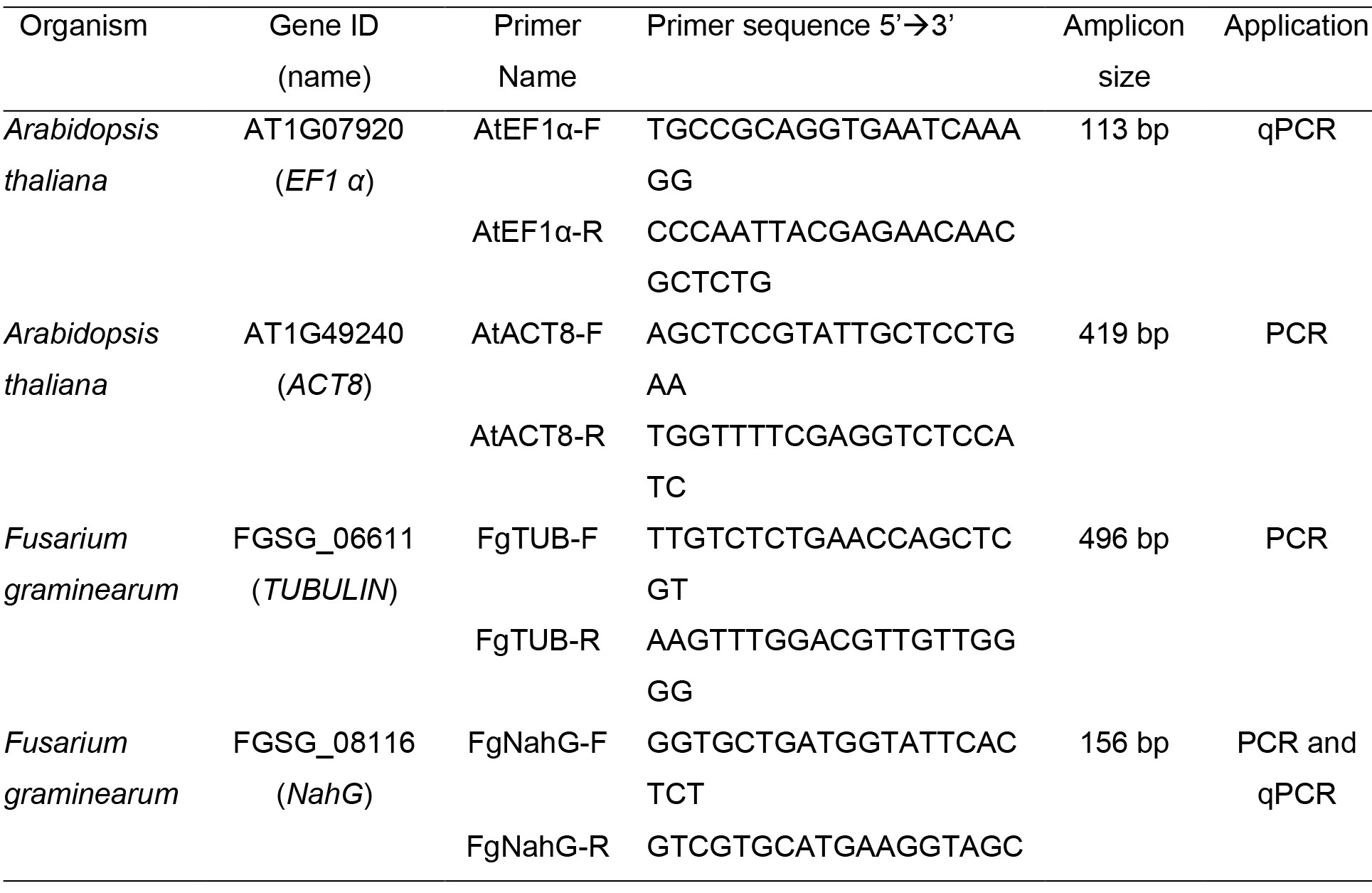
- Inoculation of Arabidopsis leaves with Fusarium graminearum mycelial fragments
- Disease evaluation of Arabidopsis inflorescence tissue infected with Fusarium graminearum
- Plant Inoculation
- Select flowering plants for inoculation i.e., choose plants that possess an unbranched bolt with both open flowers on the terminal inflorescence and two to three developing siliques. It is important to include the appropriate control genotypes with each experiment.
Note: A minimum of ten plants should be chosen for inoculation. A similar number of control plants are sprayed with water. - With a permanent black marker, Mark the position on the flower stem above which only open flowers are present and below which siliques have begun developing.
Note: In order to minimize experimental error set up the plants in a randomized block design. - Spray spore suspension (1 x 105 spores/ml in sterile ddH2O water containing 0.001% Silwet L-77) using a hand-held atomizer until droplet run-off has commenced.
- After this, re-inoculate each inflorescence with inoculum dispensed from the same sprayer (four sprays per flower head).
- Control plants are inoculated in the same way using de-ionized water containing 0.001% Silwet L-77.
- The inoculated plants were covered with a transparent plastic bag to ensure high humidity and placed in a plant growth chamber set at 22 °C under a 14 h light (80-100 μE m-2 sec-1)/10 h dark regime. Three days later the plastic bag was removed and the plants left in the growth chamber for an additional 4 days.
- Select flowering plants for inoculation i.e., choose plants that possess an unbranched bolt with both open flowers on the terminal inflorescence and two to three developing siliques. It is important to include the appropriate control genotypes with each experiment.
- Fusarium-Arabidopsis Disease (FAD) score
- Disease symptoms on individual inflorescence were monitored from day 3 onwards with the final disease score taken at 7 days post inoculation. However, if the progression of disease is rapid then the final disease score can be taken on day 5 or 6.
- A numerical scoring system developed by Urban et al. (2002) (Table 2) was used to obtain the Fusarium-Arabidopsis Disease (FAD) score.
- Disease phenotypes are assessed for three separate floral subcomponents (i) Flowers (F): infection covering open flowers and buds. (ii) New siliques (NS): Infection severity on siliques that developed after inoculation from flowers that were fully open at the time of inoculation. These flowers were located above the permanent mark placed on the stem at the time of fungal inoculation. (iii) Older siliques (OS): infection severity on siliques that existed at the time of fungal inoculation. For each of these components, the severity of infection is classified based on the macroscopic assessment of symptoms (Figure 10), as denoted in Table 2.
- The final Fusarium-Arabidopsis disease (FAD) value is calculated by addition of the three subcomponent scores, i.e., F + NS + OS = FAD as described by Urban et al. (2002). A representative data set is presented in Table 3.
- Arabidopsis genotypes with FAD values of 3 and below are classified as exhibiting resistance to Fg whereas those with values of 10 and above are classified as susceptible.
Table 2. Classification of disease phenotypes and scores for flower, and new and old siliques. Adapted from Urban et al. (2002).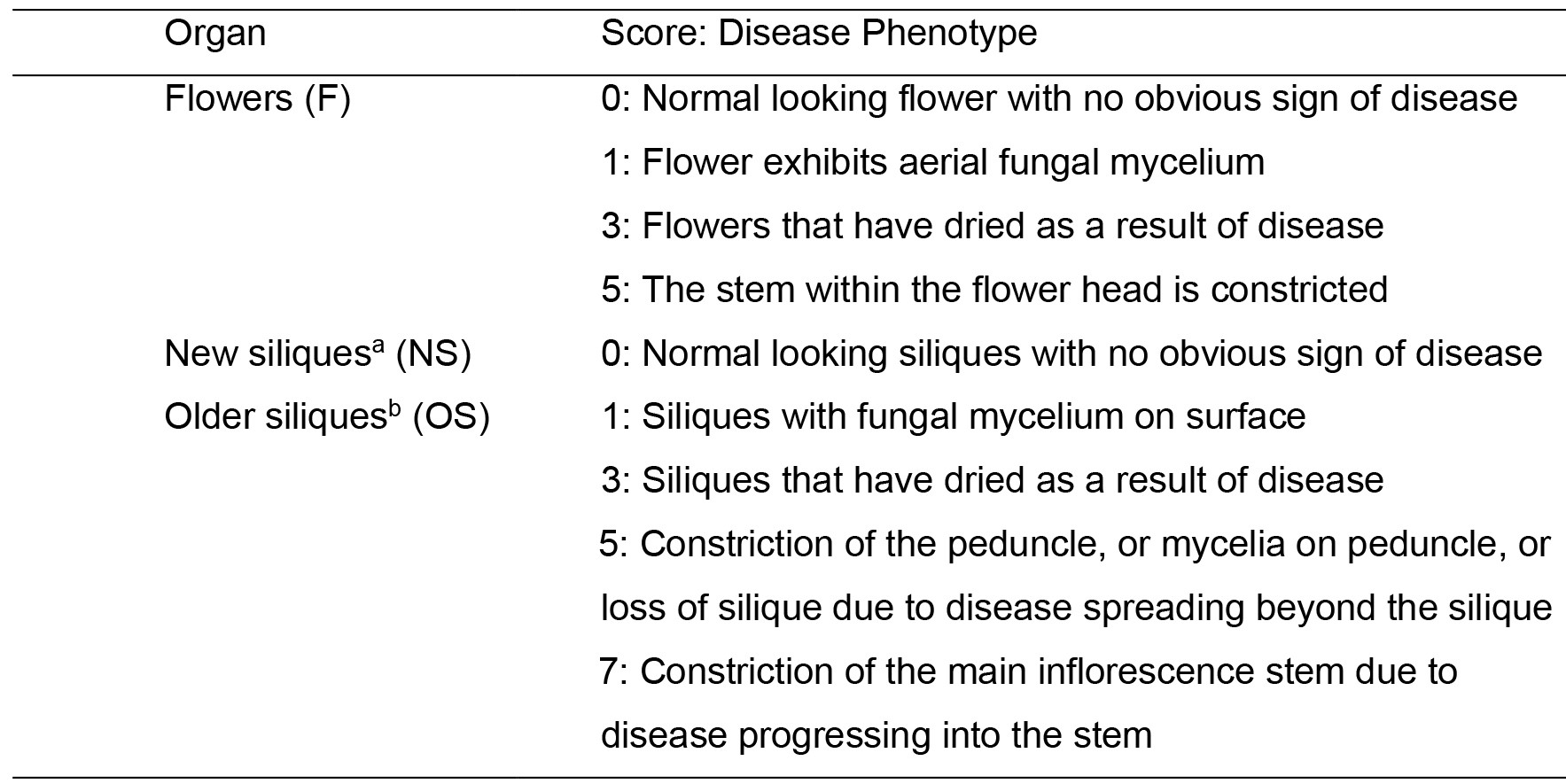
aSiliques formed during the seven day period after fungal inoculation.
bSiliques that were present when plants were sprayed with fungal macroconidia
Figure 10. Symptoms of Fusarium graminearum disease in Arabidopsis inflorescence. A. A susceptible inflorescence showing disease symptoms. B. A relatively resistant inflorescence showing production of new flowers and siliques developed from flowers that were present seven days earlier at the time of fungal macroconidia inoculation.
Table 3. Fusarium graminearum disease on inflorescence of Arabidopsis WT accession Col-0 and the mut-1 and mut-2 mutants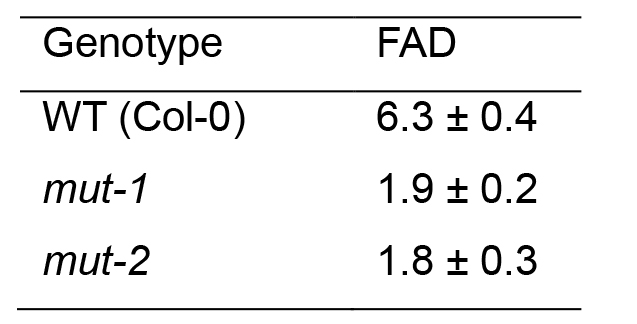
- Disease symptoms on individual inflorescence were monitored from day 3 onwards with the final disease score taken at 7 days post inoculation. However, if the progression of disease is rapid then the final disease score can be taken on day 5 or 6.
- Plant Inoculation
Recipes
- Potato Dextrose Agar-Half strength (½ PDA)
Potato dextrose broth powder 19.5 g
Agar 7.0 g
Distilled Water 1,000 ml
Adjust pH to 5.6 ± 0.2 at 25 °C, prior to adding Agar.
Sterilize by autoclaving for 20 min. Pour and allow to set approximately 30 ml into each 100 x 15 mm petri dish. - Carboxymethyl Cellulose (CMC) media
NH4NO3 1.0 g
KCl 0.2 g
MgSO4·7H2O 1.0 g
Yeast extract 1.0 g
Carboxymethyl cellulose 26.0 g
Distilled water to 1,000 ml
Sterilize by autoclaving for 20 min. - Arabidopsis DNA extraction buffer
200 mM Tris-Cl, pH 7.5 (Sambrook et al., 1989)
250 mM NaCl
25 mM EDTA, pH 7.5 (Sambrook et al., 1989)
0.5% SDS - Tris-equilibrated phenol-chloroform (Sambrook et al., 1989)
- Spray spore suspension
1 x 105 spores/ml in sterile ddH2O water containing 0.001% Silwet L-77
Acknowledgments
This work was supported by funding from: the U.S. Department of Agriculture (Agreement #59-0200-3-003 and 59-0790-8-060) as cooperative projects with the U.S. Wheat & Barley Scab Initiative. The protocols described here are based on the procedures developed and described by Makandar et al. (2010), Nalam et al. (2015) and Urban et al. (2002).
References
- Bowden, R. L. and Leslie, J. F. (1999). Sexual Recombination in Gibberella zeae. Phytopathology 89(2): 182-188.
- Livak, K. J. and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25(4): 402-408.
- Makandar, R., Nalam, V., Chaturvedi, R., Jeannotte, R., Sparks, A. A. and Shah, J. (2010). Involvement of salicylate and jasmonate signaling pathways in Arabidopsis interaction with Fusarium graminearum. Mol Plant Microbe Interact 23(7): 861-870.
- Nalam, V. J., Alam, S., Keereetaweep, J., Venables, B., Burdan, D., Lee, H., Trick, H. N., Sarowar, S., Makandar, R. and Shah, J. (2015). Facilitation of Fusarium graminearum infection by 9-Lipoxygenases in Arabidopsis and Wheat. Mol Plant Microbe Interact 28(10): 1142-1152.
- Sambrook, J., Fritch, E. F. and Maniatis, T. (1989). Molecular Cloning: a laboratory manual. Cold Spring Harbor Laboratory.
- Urban, M., Daniels, S., Mott, E., and Hammond-Kosack, K. (2002). Arabidopsis is susceptible to the cereal ear blight fungal pathogens Fusarium graminearum and Fusarium culmorum. Plant J 32: 961-973.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Nalam, V., Sarowar, S. and Shah, J. (2016). Establishment of a Fusarium graminearum Infection Model in Arabidopsis thaliana Leaves and Floral Tissues. Bio-protocol 6(14): e1877. DOI: 10.21769/BioProtoc.1877.
Category
Plant Science > Plant immunity > Disease bioassay
Plant Science > Plant immunity > Host-microbe interactions
Microbiology > Microbe-host interactions > Fungus
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.