- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Development and Application of a Fully Blind Flexible Peptide-protein Docking Protocol, pepATTRACT


Published: Vol 6, Iss 11, Jun 5, 2016 DOI: 10.21769/BioProtoc.1831 Views: 8817
Reviewed by: Arsalan DaudiPrashanth SuravajhalaMichael Tscherner
Original research article
The authors used this protocol in:

Aug 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Peptide-mediated interactions are involved in many signaling and regulatory pathways as well as the DNA replication machinery and are linked to many pathological disorders. Many research groups are currently working towards a more detailed understanding of these important interactions by characterizing the 3D complex structures with experimental methods like X-ray crystallography and NMR. However, for a large number of peptide-protein complexes such atomistic structural information is lacking to date. Computational peptide docking methods can yield information complementary to experimental information by predicting the protein-peptide complex structure from the 3D structure of the protein and the peptide sequence. This approach can also be used to study interactions between folded and disordered proteins/protein regions (e.g., the interactions of the disordered regions in tumor suppressor p53 with its different partners). Here, we describe the development and usage of the fully blind, flexible peptide-protein docking protocol pepATTRACT. The ATTRACT docking engine is implemented as a suite of command line tools and options that can be combined at will. Therefore, ATTRACT protocols like pepATTRACT are typically invoked via a custom, hand-written shell script. Although this approach is very flexible, it limits the accessibility of ATTRACT to expert users only. To make pepATTRACT easily accessible to non-expert users, we created a web-interface which helps the user set up a peptide docking protocol by editing parameters in a web browser (www.attract.ph.tum.de/peptide.html). pepATTRACT docking scripts can then executed on the user's local machine, once the ATTRACT software has been installed. Here, we describe all the steps necessary for setting up a pepATTRACT docking run via the web-interface including installation of the ATTRACT software.
Keywords: Protein-peptide interactionMaterials and Reagents
- Atomic 3D structure of protein of interest in PDB file format (www.pdb.org)
- Sequence of peptide of interest in one-letter code
Note: The protocol was tested on peptide lengths of up to 15 residues. - Optional: information on protein residues involved in binding (literature research)
Equipment
- Any computer with Unix-based OS (Linux/Mac) and at least 2-3 GB RAM
Note: It should be sufficient to run the protocol.
Software
- ATTRACT software
- ATTRACT source code (available at www.attract.ph.tum.de/services/ATTRACT/attract.tgz)
- ATTRACT virtual machine (VM) ( www.attract.ph.tum.de/services/ATTRACT/ATTRACT.vdi.gz)
- Molecular viewer (PyMOL, VMD, Rasmol etc.)
- VirtualBox (www.virtualbox.org)
Procedure
Predicting the structure of a peptide-protein complex with the fully blind docking protocol pepATTRACT requires the following steps:
- Installing the ATTRACT software (www.attract.ph.tum.de/services/ATTRACT/attract.tgz) and a molecular viewer.
- Obtaining the protein structure and the sequence of the peptide of interest.
- Generating a docking script and docking files with the pepATTRACT web interface (www.attract.ph.tum.de/peptide.html).
- Running the docking script on the user’s local machine.
Instructions and the main parts of the pepATTRACT docking protocol are visualized in Figure 1.
In the following, the individual steps are described in more detail.
- Installation of the ATTRACT software
The user can choose between building and installing ATTRACT directly from the source code or downloading an ATTRACT virtual machine (VM). The virtual machine has ATTRACT and all its dependencies installed. Note that ATTRACT can only be installed on Unix-based OS (Linux/Mac). The ATTRACT VM can be used on a large number of operating systems including Windows, Linux, Macintosh and Solaris (the following instructions for the ATTRACT VM are valid for all operating systems).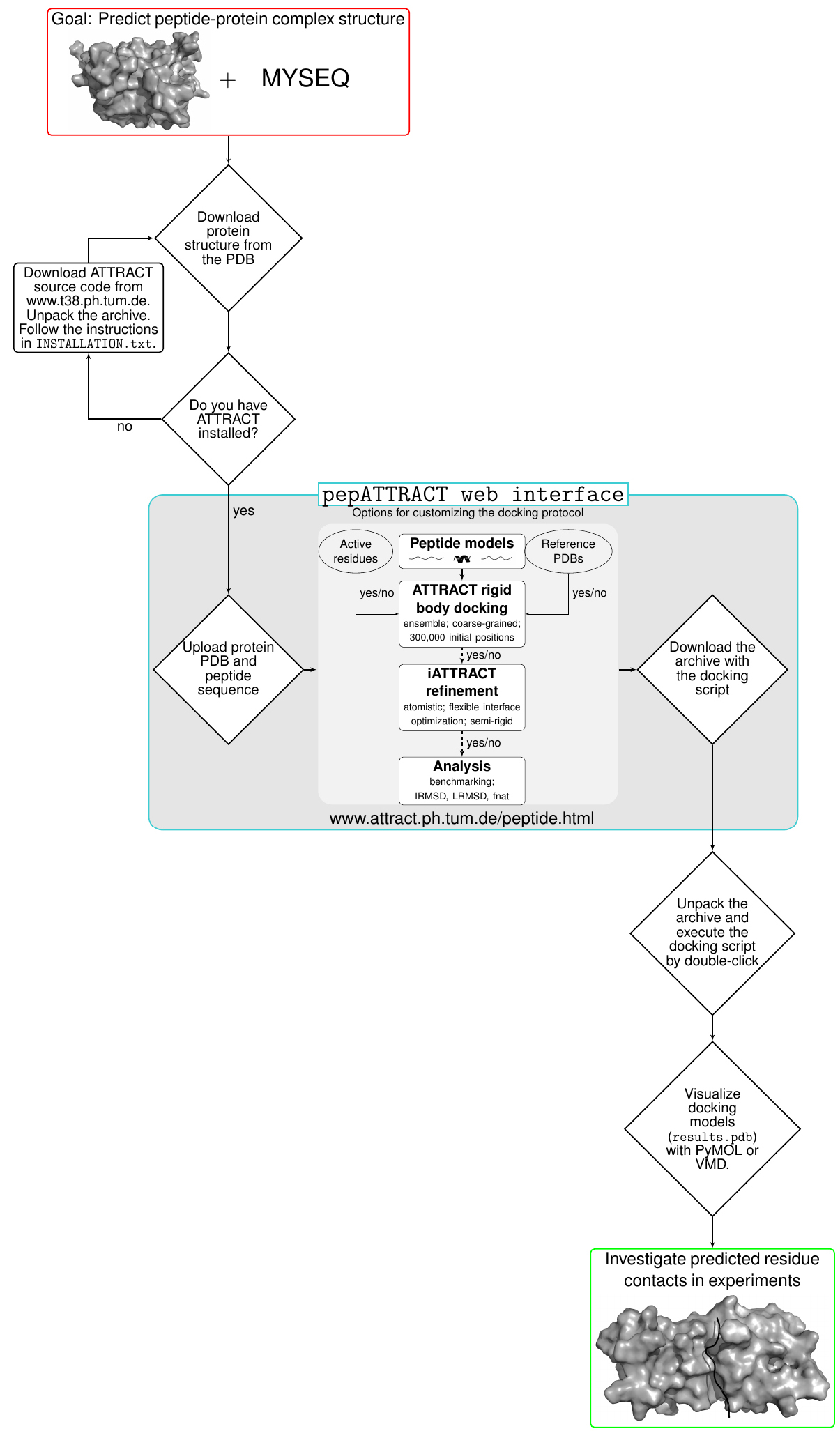
Figure 1. A flow chart illustrating the usage of the pepATTRACT docking protocol and its web interface - Instructions for ATTRACT VM
- Download and install VirtualBox (www.virtualbox.org), any version can be used.
- Download the ATTRACT VM from www.attract.ph.tum.de/services/ATTRACT/ATTRACT.vdi.gz and unpack the file.
- Open the VirtualBox program. Add the ATTRACT.vdi file to VirtualBox (click the “New” button, pick the option “Use an existing virtual hard disk file” and select the ATTRACT.vdi file, follow the instructions). Start the virtual machine to use ATTRACT.
- Download and install VirtualBox (www.virtualbox.org), any version can be used.
- Instructions for installing ATTRACT from source code
Example commands are given for Ubuntu OS 14.04.- Download the ATTRACT source code from www.attract.ph.tum.de/services/ATTRACT/attract.tgz.
- Open a terminal and unpack the source code (tar xzf attract.tgz)
- Install g++ and gfortran (sudo apt-get install g++ gfortran)
- Install numpy and scipy (sudo apt-get install python-numpy python-scipy)
- Install pdb2pqr (sudo apt-get install pdb2pqr)
- Go into attract/bin, type make clean and then make all or make all -j 4 (if you have 4 cores)
- Edit your .bashrc file and add the following lines to it:
- Export ATTRACTDIR=/home/yourname/attract/bin *(i.e. wherever you installed attract)*
- Export ATTRACTTOOLS=$ATTRACTDIR/../tools
- Export PYTHONPATH=$PYTHONPATH:/usr/share/pdb2pqr
- Type source ~/.bashrc
- Download the ATTRACT source code from www.attract.ph.tum.de/services/ATTRACT/attract.tgz.
- Installation of a molecular viewer
The molecular viewer PyMOL can be installed on Ubuntu by entering sudo apt-get install pymol in the terminal. Open a PDB file with it by typing pymol myprotein.pdb. - Input preparation
The user has to supply an atomic 3D structure of the protein of interest in PDB file format.
Alternatively, good homology models with sufficient sequence similarity can be used. Using a sequence alignment, such models can be built with programs like MODELLER (Eswar et al., 2014). Homology models can also be found in public databases; e.g., ModBase (modbase.compbio.ucsf.edu/modbase-cgi/index.cgi), the Protein Model Portal (www.proteinmodelportal.org) or the Protein Model Data Base (bioinformatics.cineca.it/PMDB). Investigations on structural deviations of homology models have shown that good models can be generated for sequence identities of e.g., > 40% (accuracy statistics of EVA at pdg.cnb.uam.es/eva/cm/res/accuracy.html; Eyrich et al., 2001; Rodrigues et al., 2013). Note that the docking protocol is quite sensitive to structural changes of the docking partners and hence it is sensitive to structural deviations in the input structure resulting from incorrect homology modeling. At the moment, unnatural and modified amino acids (e.g., post-translational modifications) are not supported and have to be removed from the input PDB file or mutated to standard amino acids manually (see Note 2). We are working towards adding support for modified amino acids to pepATTRACT in the next ATTRACT release. - Generating the docking script and executing it
- Go to www.attract.ph.tum.de/peptide.html (Figure 2).
- Upload the PDB file of the protein.
- Enter the sequence of the peptide in one-letter code (standard amino acids only).
- Specify optional parameters (see next paragraphs for details).
- Hit the “Get configuration” button.
- Download the archive yourrunname.tgz and unpack it.
- Run the protocol by double-click (typical run time 1-4 h).
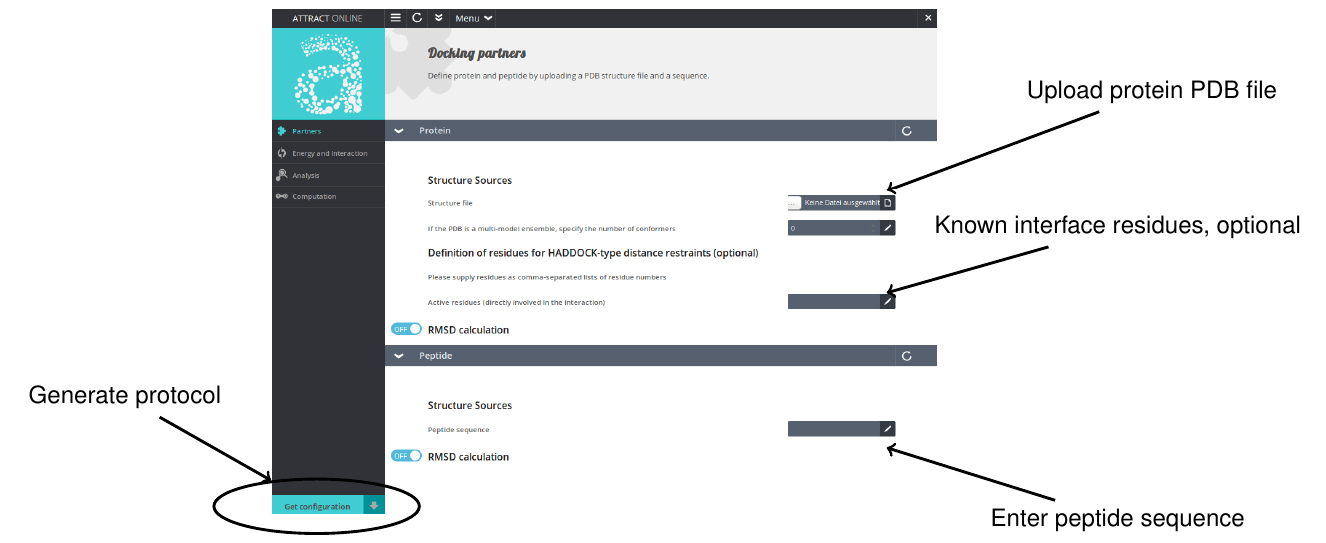
Figure 2. Screenshot of the pepATTRACT web interface with instructions
- Go to www.attract.ph.tum.de/peptide.html (Figure 2).
- Including experimental information
The web interface offers the possibility to include experimental information in the docking run and restrict the search for the peptide binding site to a portion of the protein’s surface. If certain protein residues are known to be important in peptide binding e.g., from mutational experiments, they can be specified as active residues. This will ensure that only solutions in which these residues are in contact with the peptide are generated. Multiple residues can be specified as active residues separated by commas. There is no limit on the number of possible active residues, however, keep in mind that the more residues are specified the less specific the search gets while increasing the computational load. - Protein conformational change
Many proteins undergo conformational changes when binding to a partner molecule. Although this is less common for peptide-protein than for protein-protein interactions, it influences the prediction quality of this semi-rigid peptide docking approach strongly. The pepATTRACT protocol allows to include conformational change on the protein side by providing an ensemble of possible conformations. This option is also useful if the protein structure has been derived from template-based homology modeling. Instead of uploading a PDB file containing a single protein structure to the web interface, the user can upload a multi-model PDB file and then has to specify the number of conformers present in the uploaded multi-model PDB file (web interface field “if the PDB is a multi-model ensemble, specify the number of conformers”). In a multi-model PDB file, different structures of the protein are separated by MODEL and ENDMDL lines. For more information on the format please visit deposit.rcsb.org/adit/docs/pdb_atom_format.html - MODEL. Please note that all the conformers need to have the same sequence and coordinate information for the same residues in the same order. Unfortunately, predicting conformational change upon binding is very difficult due to the large number of degrees of freedoms involved. Protein flexibility can be tested to a certain degree by performing molecular dynamics simulations or normal mode calculations prior to docking. - Analysis
The final models are converted to PDB file format for visual inspection by the user (type pymol results.pdb to look at the structures). The final structures could then be further refined in molecular dynamics simulations (Schindler et al., 2015a). The web interface offers the opportunity to benchmark pepATTRACT’s performance by docking previously experimentally resolved peptide-protein complexes and evaluating how accurate the experimental structure can be reproduced. For this, reference PDB files containing the protein structure and the peptide structure can be uploaded. Standard evaluation criteria like ligand-RMSD, interface-RMSD and fraction of native contacts (Mendez et al., 2005) can be selected for automatic computation (results.lrmsd, results.irmsd, results.fnat) (see also Note 1). - Docking protocol description
The web interface generates an archive containing the PDB files and a bash script (yourruname.sh). The bash script contains all the commands necessary to run the fully blind peptide-protein docking protocol pepATTRACT for a given protein structure and peptide sequence. The docking protocol consists of the following steps (Figure 1). First, three peptide model structures are generated from sequence (α-helical, extended and poly-proline conformation) (Tien et al., 2013). This approach is supported by the experimental observation that this limited set of three motifs dominates the conformational ensemble that peptides adopt in peptide-protein complexes (London et al., 2010) and has also been successfully used in the HADDOCK peptide docking protocol (Trellet et al., 2013). Then global rigid body docking with ATTRACT using a coarse-grained force field is performed (Zacharias, 2003; May and Zacharias, 2008). The ATTRACT coarse-grained force field uses soft distance-dependent Lennard-Jones (LJ)-type potentials with attractive or repulsive parameters and electrostatics between charged residues to calculate the interaction energy between residue types A and B.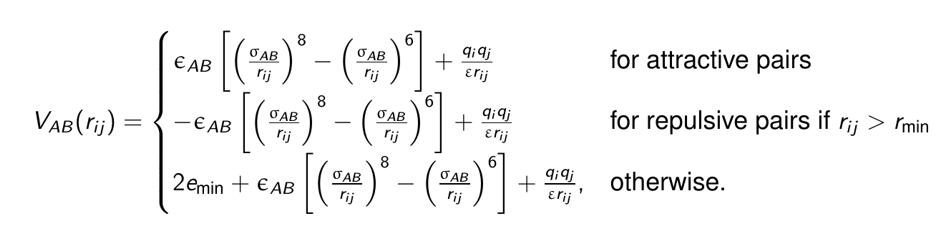
Notes:- The ATTRACT energy/score is only used to compare/rank different docking models, it does not represent a measure for peptide binding affinity.
- In the coarse-grained docking stage, the internal structure of the protein and the peptide are kept rigid and only orientational and translational degrees of freedoms are explored (6 rigid body degrees of freedom).
- The energy calculations are accelerated by a precalculated grid (de Vries et al., 2015). The rigid body docking solutions are rescored and ranked by ATTRACT score.
- The top ranked models can then be refined with the flexible interface refinement method iATTRACT (Schindler et al., 2015b).
- In the refinement stage, the models are subjected to a simultaneous optimization of their global rigid body degrees of freedom and of the local position of the interface atoms.
- The ATTRACT energy/score is only used to compare/rank different docking models, it does not represent a measure for peptide binding affinity.
Representative data
As an example application, we predict the complex of a peptide derived from type 1 human immunodeficiency virus (HIV-1) capsid protein with a cellular protein, cyclophilin A (CypA). The HIV-1 virion forms by assembly of the Gag polyprotein. Approximately 2,000 Gag molecules bind to the host cell membrane and assemble into budding virions. The HIV-1 virion also contains about 200 copies of the cytosolic CypA protein. These are essential for virus replication and are packaged into the virion by a direct interaction between Gag and CypA. Previous studies have characterized both the 3D complex structure of CypA and the His87-Ala-Gly-Pro-Ile-Ala92 sequence from the capsid protein (PDB code: 1AWR) (Vajdos et al., 1997) and the apo cypA structure (PDB code: 2ALF). We use these structures to test the pepATTRACT protocol refining the top 1,000 rigid body docking models with iATTRACT. One of the final top 10 ranked models is shown in Figure 3. It correctly predicts the binding site and an extended conformation for the peptide (IRMSD 1.15 A°, fraction of native contacts 0.67).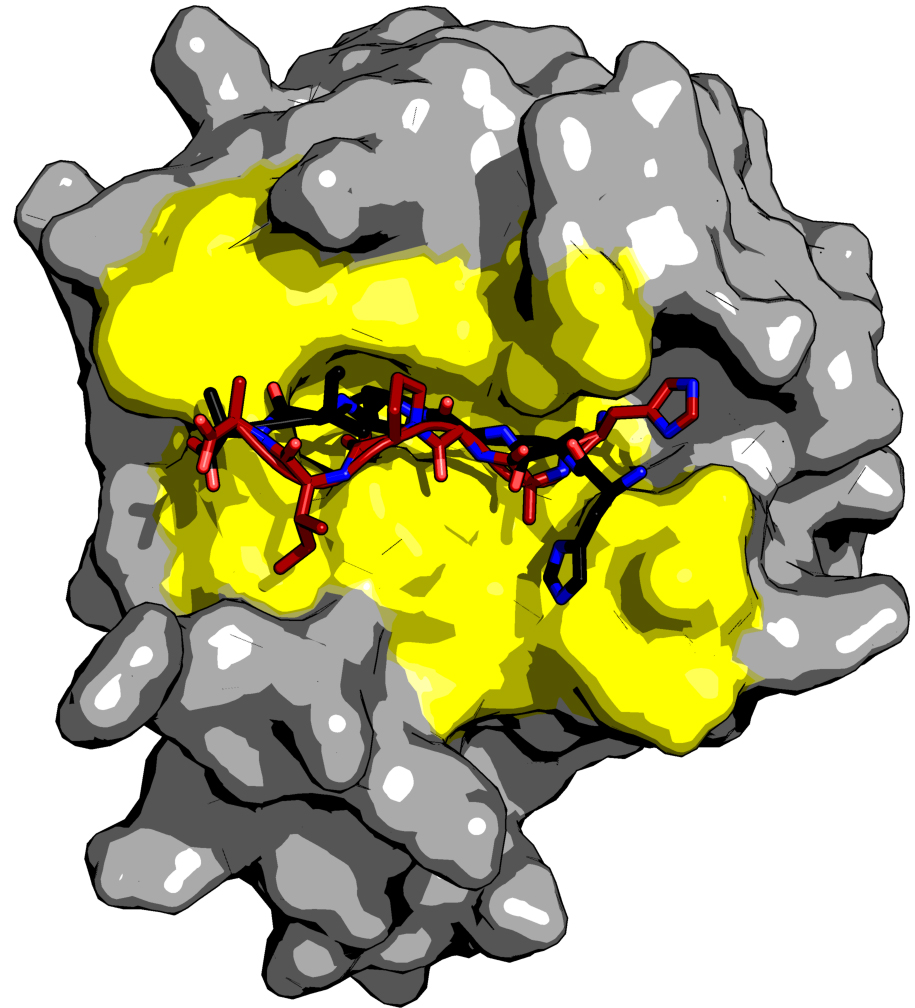
Figure 3. Results for docking cyclophilin A to the HAGPIA sequence of HIV-1 capsid protein. The protein is drawn in surface representation (gray), the peptide in stick representation (red: docking model, black: reference from the crystal structure). The protein residues involved in binding are shown in yellow. This figure was generated with PyMOL.
Notes
- Performance
pepATTRACT’s performance has been benchmarked on a set of 80 known peptide-protein complexes (Schindler et al., 2015a) yielding an overall success rate of 70% for 1,000 generated final docking models per test case. pepATTRACT was also compared to two state-of-the-art local docking methods, Rosetta FlexPepDock ab-initio (Raveh et al., 2011) and HADDOCK peptide docking (Trellet et al., 2013). We found that pepATTRACT's performance in fully blind mode was comparable to that of the two local docking methods and pepATTRACT-local surpassed their performance by a significant margin (Schindler et al., 2015a). - Reproducibility
Nevertheless, we recommend the users test the protocol for their specific biological systems whenever possible using similar experimentally resolved complex structures as benchmark cases. Rerunning the protocol with identical input files on the same machine will give identical results. However, due to different compiler settings and numerical instabilities, executing the same commands on different machines will lead to slightly different numerical results. These slight differences are not significant in terms of overall prediction quality (i.e., the success rates for testing the protocol on a benchmark set of known peptide-protein complexes). - Cofactors and modified amino acids
The ATTRACT docking engine does not support ions, cofactors or modified amino acids at the moment, we are working towards including these in future versions of ATTRACT. Currently, all HETATM entries are ignored when reading the PDB file. Users should manually convert modified amino acids to standard amino acids before uploading their PDB file and relabel them to ATOM entries. - Memory requirements
For running the protocol, a precalculated potential energy grid has to be loaded into memory. This requires at least 2 GB of RAM, for larger proteins, the demand can be higher. Failures of the protocol may result from failure to allocate sufficient memory. - Large protein
The ATTRACT software is compiled with default settings which limits the number of atoms in a protein to 10,000. This limit can be enlarged by modifying the file $ATTRACTDIR/max.h and increasing the variable MAXATOM (maximum number of atoms in protein) and if necessary MAXRES (maximum number of residues), TOTMAXATOM, TOTMAXRES etc. to the desired range. Then recompile by going to $ATTRACTDIR and typing make clean and make all. Check the OPLS converted file yourprotein-aa.pdb to find out how many atoms the protein has during docking with ATTRACT. Keep in mind that larger proteins also imply more memory and longer run times. - Other files generated by the web interface
Apart from the .tgz docking archive, the web interface also offers two other files for download: an embedded parameter file (yourrunname-embedded.web) and a delta file (yourrunname-delta.json). The embedded parameter file contains all docking parameters needed by ATTRACT in a single file (including the uploaded protein PDB file). This file describes the docking protocol in a reproducible manner and can be used for automatic recalculation of docking runs or later modification of parameters via the Upload web-interface (www.attract.ph.tum.de/services/ATTRACT/upload.html). The delta file contains the web form parameters that were filled in by the user or changed (when uploading a previously generated embedded parameter file). The delta file is the most useful reference file for describing the docking protocol in words, since it contains all the docking parameters in a text file. It can be used as a reference for writing a protocol description in the Materials & Methods section. Furthermore, the delta file should be provided in all help, support and feedback requests and for bug reports.
Acknowledgments
This protocol was adapted from the previously published studies of de Vries et al. (2015) and Schindler et al. (2015a). The authors acknowledge funding by the Center for Integrated Protein Science Munich (CIPSM). This work was performed on computational resources provided by a CIPSMWomen’s consumables grant.
References
- de Vries, S. J., Schindler, C. E., Chauvot de Beauchene, I. and Zacharias, M. (2015). A web interface for easy flexible protein-protein docking with ATTRACT. Biophys J 108(3): 462-465.
- Eyrich, V. A., Marti-Renom, M. A., Przybylski, D., Madhusudhan, M. S., Fiser, A., Pazos, F., Valencia, A., Sali, A. and Rost, B. (2001). EVA: continuous automatic evaluation of protein structure prediction servers. Bioinformatics 17(12): 1242-1243.
- London, N., Movshovitz-Attias, D. and Schueler-Furman, O. (2010). The structural basis of peptide-protein binding strategies. Structure 18(2): 188-199.
- May, A. and Zacharias, M. (2008). Energy minimization in low-frequency normal modes to efficiently allow for global flexibility during systematic protein-protein docking. Proteins 70(3): 794-809.
- Mendez, R., Leplae, R., Lensink, M. F. and Wodak, S. J. (2005). Assessment of CAPRI predictions in rounds 3-5 shows progress in docking procedures. Proteins 60(2): 150-169.
- Raveh, B., London, N., Zimmerman, L. and Schueler-Furman, O. (2011). Rosetta FlexPepDock ab-initio: simultaneous folding, docking and refinement of peptides onto their receptors. PLoS One 6(4): e18934.
- Rodrigues, J. P., Melquiond, A. S., Karaca, E., Trellet, M., van Dijk, M., van Zundert, G. C., Schmitz, C., de Vries, S. J., Bordogna, A., Bonati, L., Kastritis, P. L. and Bonvin, A. M. (2013). Defining the limits of homology modeling in information-driven protein docking. Proteins 81(12): 2119-2128.
- Schindler, C. E., de Vries, S. J. and Zacharias, M. (2015a). Fully Blind Peptide-Protein Docking with pepATTRACT. Structure 23(8): 1507-1515.
- Schindler, C. E., de Vries, S. J. and Zacharias, M. (2015b). iATTRACT: simultaneous global and local interface optimization for protein-protein docking refinement. Proteins 83(2): 248-258.
- Tien, M. Z., Sydykova, D. K., Meyer, A. G. and Wilke, C. O. (2013). PeptideBuilder: A simple Python library to generate model peptides. Peer J 1: e80.
- Trellet, M., Melquiond, A. S. and Bonvin, A. M. (2013). A unified conformational selection and induced fit approach to protein-peptide docking. PLoS One 8(3): e58769.
- Vajdos, F. F., Yoo, S., Houseweart, M., Sundquist, W. I. and Hill, C. P. (1997). Crystal structure of cyclophilin A complexed with a binding site peptide from the HIV-1 capsid protein. Protein Sci 6(11): 2297-2307.
- Webb, B. and Sali, A. (2014). Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics 47: 5 6 1-32.
- Zacharias, M. (2003). Protein-protein docking with a reduced protein model accounting for side-chain flexibility. Protein Sci 12(6): 1271-1282.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Schindler, C. E., de Vries, S. J. and Zacharias, M. (2016). Development and Application of a Fully Blind Flexible Peptide-protein Docking Protocol, pepATTRACT. Bio-protocol 6(11): e1831. DOI: 10.21769/BioProtoc.1831.
Category
Biochemistry > Protein > Structure
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.