- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vitro Microtubule Binding Assay and Dissociation Constant Estimation
Published: Vol 6, Iss 6, Mar 20, 2016 DOI: 10.21769/BioProtoc.1759 Views: 15793
Reviewed by: Arsalan DaudiMarta BjornsonTamara Vellosillo
Original research article
The authors used this protocol in:

Sep 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Microtubules (MTs) support an astonishing set of versatile cellular functions ranging from cell division, vesicle transport, and cell and tissue morphogenesis in various organisms. This versatility is in large mediated by MT-associated proteins (MAPs). The neuronal MAP Tau, for example, is stabilizing MTs in axons of the vertebrate nervous system and thus provides the basis for enduring axonal transport and the long life span of neurons (Mandelkow et al., 1994). Tau has been shown to bind to MTs directly in vitro and also to promote their nucleation from α-/β-tubulin subunits (Goode et al., 1994). Recently, we identified a plant-specific protein family called “companion of cellulose synthase” (CC), which was shown to bind MTs and enhance dynamics of the cortical MT array in plant cells under salt stress (Endler et al., 2015). The CCs were therefore hypothesized to help plant cells cope with stress conditions and thereby maintain biomass production under adverse growth conditions. Here, we provide detailed experimental information on in vitro MT binding assays, which allow assessing whether a protein of interest is binding to MTs. The assay utilizes the high molecular weight of MTs in a spin down approach and enables the determination of the dissociation constant Kd, a measure for the protein’s binding strength to MTs.
Materials and Reagents
- Tubes, with Snap-On Cap, Polypropylene (1.5 ml, 11 x 38 mm) Natural (Beckman Coulter, catalog number: 357448 )
Note: Any ultracentrifuge tubes can be used. Make sure the volume of the tubes does not exceed 1.5 to 2 ml. Otherwise handling of the assay is unfeasible. - PIPES (Sigma-Aldrich, catalog number: P6757 )
- Trizma® base (Sigma-Aldrich, catalog number: T1503 )
- Magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: M8266 )
- Ethylene glycol-bis(2-aminoethylether)-N, N, N’, N’-tetraacetic acid (EGTA) (Sigma-Aldrich, catalog number: E3889 )
- Glycerol (Sigma-Aldrich, catalog number: G5516 )
- Bovine Serum Albumin (BSA) (Sigma-Aldrich, catalog number: A2153 )
- Paclitaxel (Taxol) (Sigma-Aldrich, catalog number: T7402 )
- Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, catalog number: 276855 )
- Guanosine 5’-triphosphate sodium salt hydrate (GTP) (Sigma-Aldrich, catalog number: G8877 )
- Tubulin protein, bovine, lyophilized (> 99% pure) (Cytoskeleton Inc., catalog number: TL238 ) or Tubulin protein (> 99% pure): porcine brain (Cytoskeleton Inc., catalog number: TL240 )
- Microtubule-associated protein rich fraction: bovine brain (Cytoskeleton Inc., catalog number: MAPF )
- NuPAGE Novex 4-12% Bis-Tris Protein Gels, 1.0 mm, 15-well (Thermo Fisher Scientific, catalog number: NP0323PK2 )
Note: Any other 1D protein gel able to separate tubulin and the protein of interest works as well. - Brilliant Blue G (Sigma-Aldrich, catalog number: 27815 )
- Aluminum sulfate-(14-18)-hydrate (Sigma-Aldrich, catalog number: 368458 )
- 85% Orthophosphoric acid (Sigma-Aldrich, catalog number: 345245 )
- Sodium dodecyl sulfate (Sigma-Aldrich, catalog number: L3771 )
- Bromophenol blue (Sigma-Aldrich, catalog number: B0126 )
- 2-mercaptoethanol (Sigma-Aldrich, catalog number: M6250 )
- Brinkley buffer 1980 (BRB80) (see Recipes)
- BRB80 Cushion buffer (see Recipes)
- Taxol stocks (see Recipes)
- GTP stocks (see Recipes)
- Tubulin stocks (see Recipes)
- MAP fraction stocks (see Recipes)
- BSA stocks (see Recipes)
- Colloidal Coomassie stain (see Recipes)
- Laemmli buffer (see Recipes)
Equipment
- Optima MAX-XP Ultracentrifuge (Beckman Coulter, catalog number: 393315 )
- TLA-55 Rotor Package, Fixed-Angle (45° Angle), Aluminium (Beckman Coulter, catalog number: 366725 )
Note: Any combination of ultracentrifuge and rotor can be used. - BIO RAD ChemiDOC MP Imaging System (Bio-Rad Laboratories, catalog number: 1708280 )
Note: A normal document scanner can be used to scan protein gels instead. The scanner needs to be able to export scans in an uncompressed format. - SDS-PAGE system to separate protein in 1D
- Vortex mixer
Software
- ImageJ (Fiji)
- Spreadsheet software (e.g., Microsoft Excel)
- GraphPad Prism Version 6.01 (or higher) (GraphPad Software Inc)
Procedure
- MT polymerization
- Defrost one taxol aliquot at room temperature (RT) (10 µl of 10 mM taxol in DMSO) and mix it with 990 µl of BRB80 (BRB80-T) (final taxol concentration = 100 µM). Keep at RT.
- Defrost one tubulin aliquot (10 µl of 4 mg/ml tubulin) in an RT water bath until thawed. Immediately transfer aliquot to ice.
- Add 1 µl of BRB80 Cushion buffer to the tubulin aliquot and incubate for 20 min at 37 °C in a water bath to polymerize MTs. We advise to use a water bath for uniform heating of the sample.
- After incubation add 100 µl of BRB80-T to the MTs to stabilize them (final tubulin concentration = approx. 4 µM). Keep at RT from now on.
- Defrost one taxol aliquot at room temperature (RT) (10 µl of 10 mM taxol in DMSO) and mix it with 990 µl of BRB80 (BRB80-T) (final taxol concentration = 100 µM). Keep at RT.
- MT binding assay
Note: The assay relies on the fact that MTs, because of their large molecular weight, pellet when spun at 100,000 x g. If a protein of interest binds to MTs, it will thus also be found in the pellet. For the assay to provide reliable results, it is necessary that the test protein is stable under the conditions used during centrifugation (check various buffers if that is not the case). We recommend testing the stability of your protein by running a centrifugation of your protein in buffer solution without adding MTs. This test ascertains that the protein is not (or only negligibly) found in the pellet in absence of MTs (please also refer to Figure 1). In general, the salt concentration in the assay should be kept minimal as high salt concentrations interfere with MT binding. In our hands, the interaction between the MTs and the CC proteins was affected at NaCl concentrations above approx. 60 mM.- Prepare 7 reactions as described in Table 1. BSA is used as a negative control as it does not bind to MTs. A mixture of different MT binding proteins (MAPF) serves as positive control. The concentration of the test protein should be high enough to add a minimum of 5 µg of protein to the assay. Fill up to a volume of 50 µl using BRB80-T. For practical guidelines on protein expression in E. coli, please refer to Sivashanmugam et al. (2009) and Rosano and Ceccarelli (2014).
Table 1. Experimental setup for the microtubule binding assay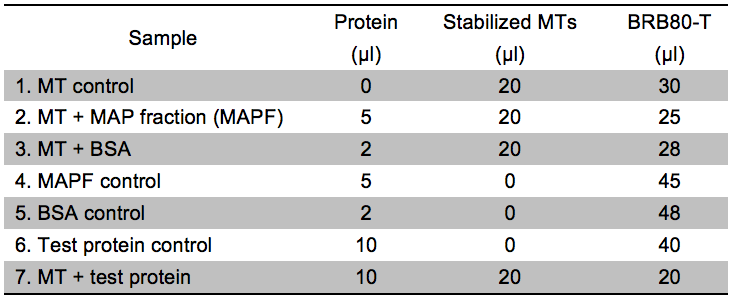
- Incubate the reactions for 30 min at RT.
- Defrost one aliquot of taxol and add 990 µl of BRB80 Cushion buffer (BRB80-CT). Mix gently by pipetting up and down without introducing air bubbles. If air bubbles are introduced, spin the buffer 30 sec at 10,000 x g.
- Prepare 7 ultracentrifuge tubes and add 100 µl of BRB80-CT to each tube. Again, avoid introducing air bubbles to the buffer.
- Place the reactions as outlined in Table 1 gently on top of the BRB80-CT without mixing the 2 solutions. By spinning the reactions through the cushion buffer, the separation of unbound MAPs from the microtubules is enhanced.
- Centrifuge for 30 min at 100,000 x g, 23 °C.
- After centrifuging, label the side of the tube facing away from the rotor centre with a pen. This is where the pellet should be found (the pellet may not be visible).
- Note that after centrifugation the phase separation between cushion and reaction is not visible anymore. Carefully remove 30 µl from the topmost part of the solution. This is the soluble fraction of unbound protein (see Figure 1). Do not discard it!
- Carefully remove and discard the rest of the solution using a pipette. Avoid touching the area marked previously to not disturb the MT pellet. Try not to leave any solution inside of the tube as this will dilute the pellet fraction. The pellet will be located at the tube side and not at the actual bottom of the tube.
- Add 60 µl 1x Laemmli buffer (Laemmli, 1970) directly to the pellet. Mix and resuspend by vortexing.
- Add 6 µl of 5x Laemmli buffer to the supernatant fraction. Mix by vortexing.
- Separate 10 µl of each fraction by SDS PAGE and stain the gels with colloidal Coomassie stain (Dyballa and Metzger, 2009) and image the gels. See Figure 1 for representative results.
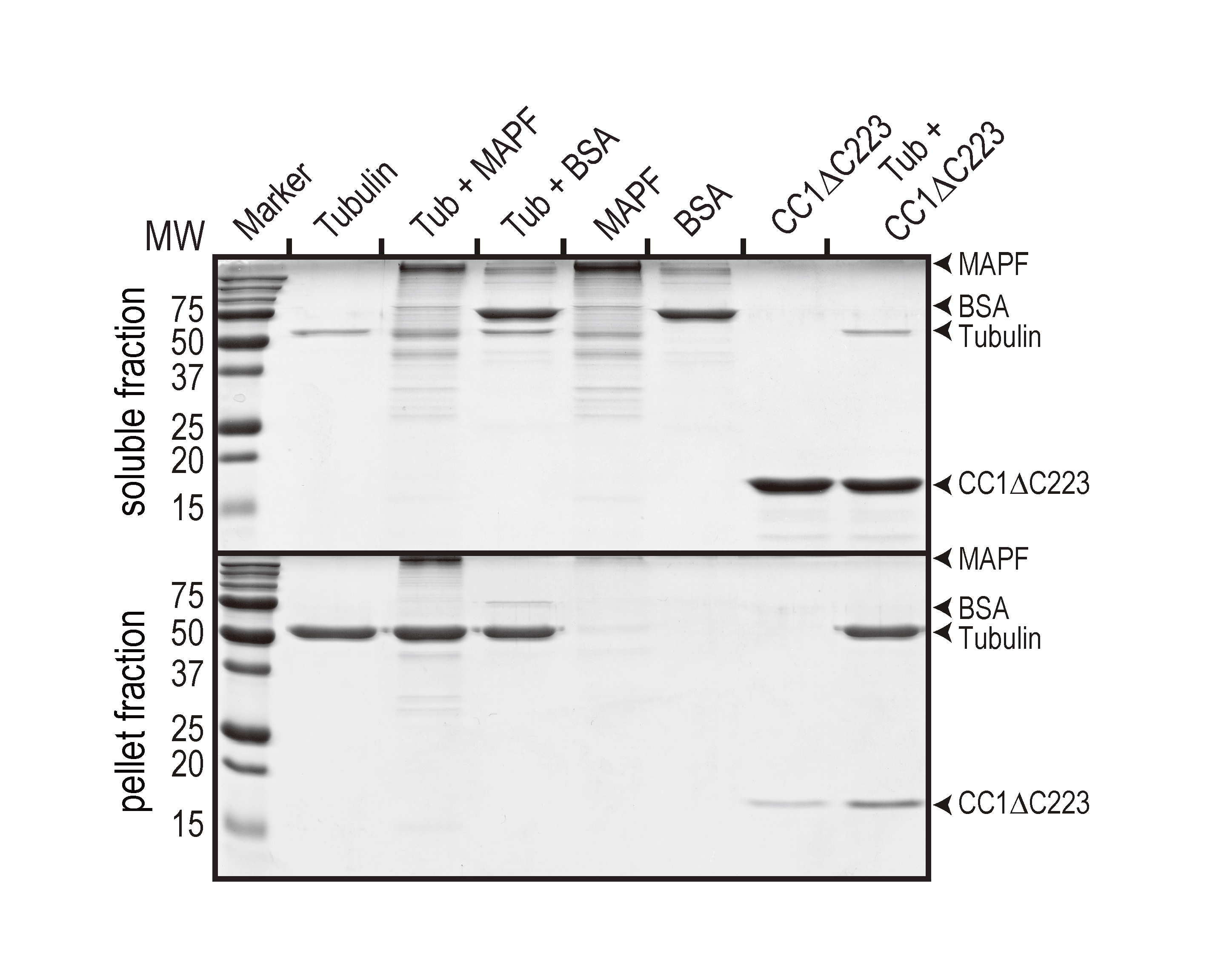
Figure 1. Representative data showing a MT binding assay. The test protein is the His-tagged, cytosolic domain of CC1 (CC1∆C223). A MAP fraction (MAPF) was used as the positive and BSA was used as the negative control, respectively. In comparison to the control without MTs (second last lane), CC1∆C223 was significantly enriched in the pellet fraction when MTs were present (last lane). The figure was taken from Endler et al. (2015).
- Prepare 7 reactions as described in Table 1. BSA is used as a negative control as it does not bind to MTs. A mixture of different MT binding proteins (MAPF) serves as positive control. The concentration of the test protein should be high enough to add a minimum of 5 µg of protein to the assay. Fill up to a volume of 50 µl using BRB80-T. For practical guidelines on protein expression in E. coli, please refer to Sivashanmugam et al. (2009) and Rosano and Ceccarelli (2014).
- Dissociation constant (Kd) estimation of an MT binding protein by gel densitometry
Note: For this assay one assumes that the underlying binding mechanism of the protein to MTs follows a one-to-one stoichiometry, i.e. one molecule of protein binds to one α-/β-tubulin subunit. If this is not the case for the protein of interest, the calculated ratio of protein bound to tubulin will eventually exceed 1:1 and the presented method is, in this case, not valid. Furthermore, the reliability of the assay depends on the MTs being stable throughout the whole experiment. If your protein shows depolymerizing or severing activity on MTs, this assay is also not appropriate (the more protein one adds, the more MTs will depolymerize or be severed, respectively).- Polymerize MTs (as described in part A).
- Prepare 6 different dilutions of the test protein with the same buffer conditions, not exceeding a volume of 30 µl. For example, for dilutions with a total volume of V = 10 µl, use x µl of test protein and add 10-x µl of buffer to maintain a stable salt concentration over all dilutions.
Note: If a volume of 20 µl is exceeded taxol may be added to a concentration of 100 µM to prevent MT depolymerisation. - Replicate the sample with the highest protein concentration as a control.
- Prepare 8 reactions as described in Table 2. It is essential that the final assays have the same conditions (see step C2).
Note: To estimate the Kd accurately, the reaction has to reach saturation, i.e., an increase in protein concentration does not lead to an increase in MT binding. Firstly, we recommend performing several spin down assays with variable concentrations of the test protein to find the point of saturation. After determination of the saturation point, it is important to collect enough data points below the saturation point to accurately fit the curve and calculate the Kd. It is good practice, to repeat the experiment (at least) 3 times to acquire enough data points for robust analysis.
Table 2. Experimental setup for the Kd estimation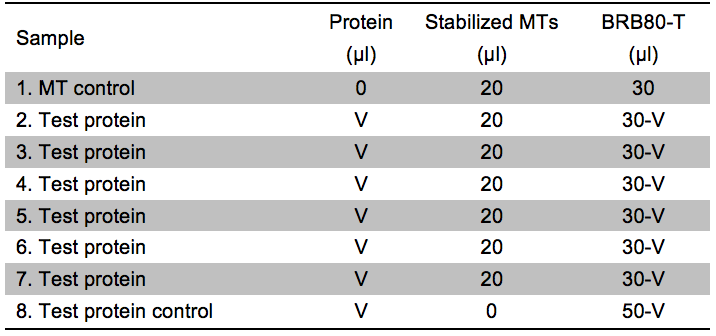
- Incubate the reactions for 30 min at RT.
- Defrost one aliquot of taxol at RT and add 990 µl of BRB80 Cushion buffer. Mix gently by pipetting up and down without introducing air bubbles. If air bubbles are introduced, spin the buffer 30 sec at 10,000 x g.
- Prepare 8 ultracentrifuge tubes and add 100 µl of BRB80-CT to each tube. Again, avoid introducing air bubbles to the buffer.
- Place the reactions as outlined in Table 2 gently on top of the BRB80-CT without mixing the 2 solutions. By spinning the reactions through the cushion buffer one enhances separation of unbound MAPs from the microtubules.
- Centrifuge for 30 min at 100,000 x g, 23 °C.
- After centrifuging, label the side of the tube facing away from the rotor centre with a pen. This is where the pellet should be found (the pellet may not be visible).
- Note that after centrifugation the phase separation between cushion and reaction is not visible anymore. Carefully remove 30 µl from the topmost part of the solution. This is the soluble fraction of unbound protein (see Figure 1). Do not discard it!
- Carefully remove and discard the rest of the solution using a pipette. Avoid touching the area marked previously to not disturb the MT pellet. Try not to leave any solution inside of the tube as this will dilute the pellet fraction. The pellet will be located at the tube side and not at the actual bottom of the tube.
- Add 60 µl 1x Laemmli buffer (Laemmli, 1970) directly to the pellet. Mix and resuspend by vortexing.
- Add 6 µl of 5x Laemmli buffer to the supernatant fraction. Mix by vortexing.
- Load 10 µl of each fraction on an SDS gel in the order shown in Figure 2A (representative results of supernatant fractions). Also load a BSA standard (5 µg) on each gel to correct for differences in staining between each gel (right-most column in Figure 2A).
- Separate the fractions by SDS page, stain the gels with colloidal Coomassie stain (Dyballa et al., 2009) and image/scan them. Save the image files in an uncompressed file format.
- Polymerize MTs (as described in part A).
Representative data and analysis
- Open the scanned gel with Fiji.
- Choose the “Rectangular Selections tool” and draw a rectangle around the first lane. The outline of the rectangle should be wide enough to cover each lane on your gel. Press “1” on the keyboard.
- A “1” will appear in the first selection. Use the mouse to click and hold inside of the first selection and drag it over to the second lane. Press “2” on your keyboard.
- A “2” will appear in the second selection. Repeat step 3 until you selected each lane on the gel with a separate rectangle. Do not forget to press “2” on the keyboard once you dropped the selection on a new lane. See Figure 2B for representative data with all lanes selected.
- Press “3” on the keyboard. Fiji will open a new window with a profile plot of each selected lane (see Figure 2C; to save space, only 4 lanes out of 9 are shown). This plot shows the relative density of each band in a lane. The lanes are arranged from top (lane 1; MT control) to bottom (lane 9; BSA standard).
- Choose the “Straight line tool” and enclose the peak right above the background signal (see Figure 2D). Make sure to enclose the peaks otherwise the next step will fail.
- Choose the “Wand tool” and click in each peak. Begin in the upper left corner (MT band from the MT control sample) and go through the all plots until you reach lane 9 (BSA standard band). A result window will open showing you the area of each peak (see Figure 2E).
- Once all peaks are analysed using the “Wand tool”, click “Analyze > Gels > Label Peaks” in Fiji. Fiji will calculate the percentage of the size of each individual peak based on the total size of all peaks in the plot (see Figure 2F).
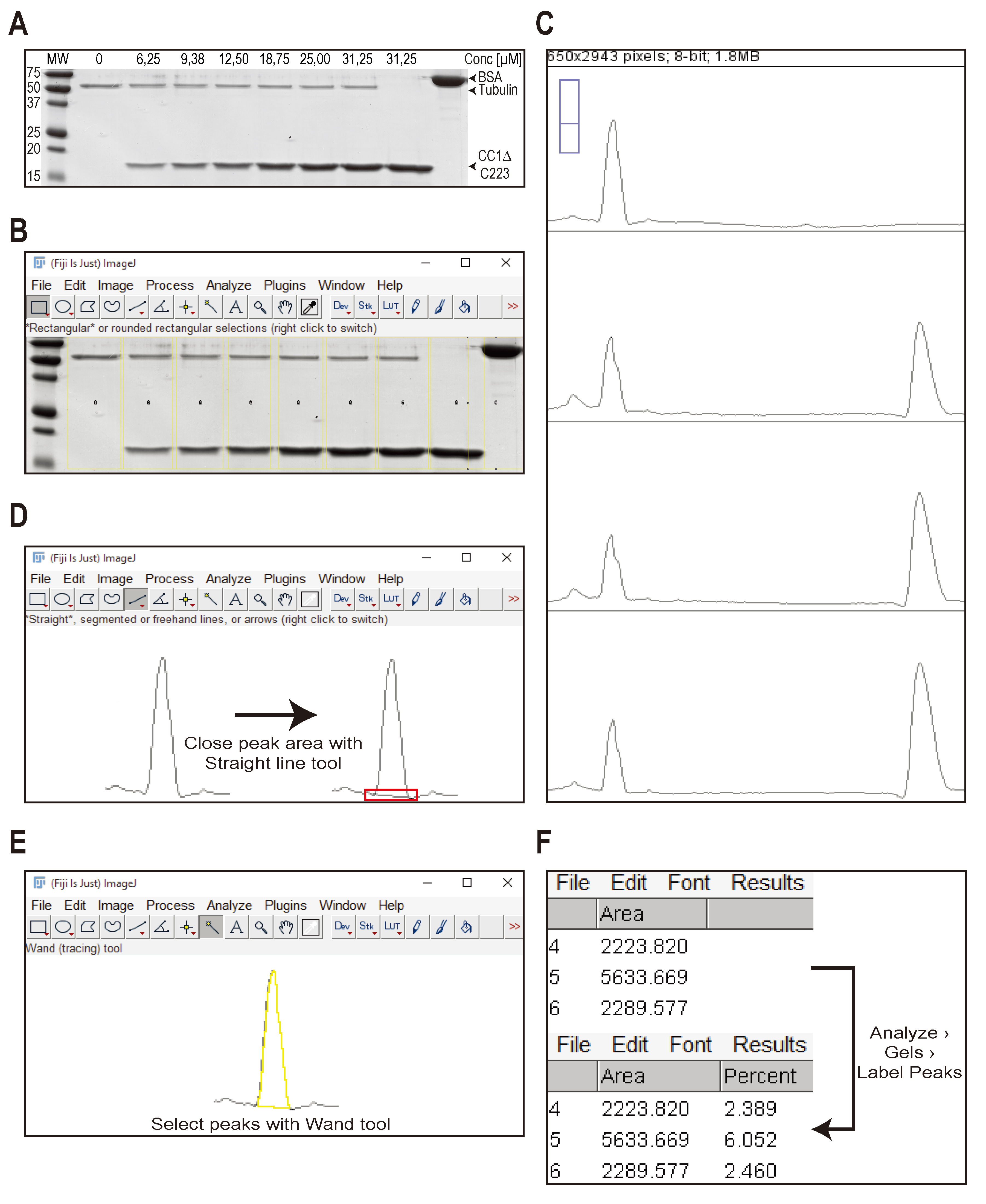
Figure 2. Workflow showing the analysis of a representative Coomassie stained gel of supernatant fractions to estimate the Kd of a MT-binding protein. A. Coomassie-stained gel showing the supernatant fraction of 8 independent MT binding experiments. Five µg of BSA is loaded on the gel as a standard (last lane). The His-tagged, cytosolic domain of CC1 (CC1∆C223) was used in the assay. Concentrations of CC1∆C223 are indicated. The molecular weights of tubulin, CC1∆C223 and BSA are indicated by arrowheads. B to F. Workflow of operations performed in Fiji to measure the peak area of the protein bands in a profile plot as described in section C, Data analysis. - Copy the data of the pellet and supernatant gels into a spreadsheet program (e.g. Microsoft Excel). Figure 3A and B show representative data to illustrate the following calculations.
- Calculate the relative density of each peak in comparison to the BSA standard on the gel (see Figure 3A).
- If the test protein is also found in the pellet in absence of MTs (test protein control, see Table 2, lane 8, and Figure 1A), we recommend quantifying it to use it for corrections later on (termed “error fraction”, see Figure 3A, red box).
- Calculate the total protein being present in each experiment by adding the values of the pellet band and the supernatant band for each protein concentration being tested (see Figure 3C, number 1).
- Correct the total protein by the error fraction.
Note: This is a simplified approach, because it assumes that the error is always the same independent from the protein concentration in the assay (see Figure 3C, number 1). - Calculate the percentage of protein bound relative to MTs using the equation highlighted in Figure 3C (number 2). If your protein alone did not migrate to the pellet, you do not have to subtract the error.
- Calculate the concentration of the test protein in the soluble fractions using the equation highlighted in Figure 3C (number 3). The protein found in the error fraction is supposed to be non-functional. Because of this, we did not add the error fraction on top of the soluble fraction of each experiment.
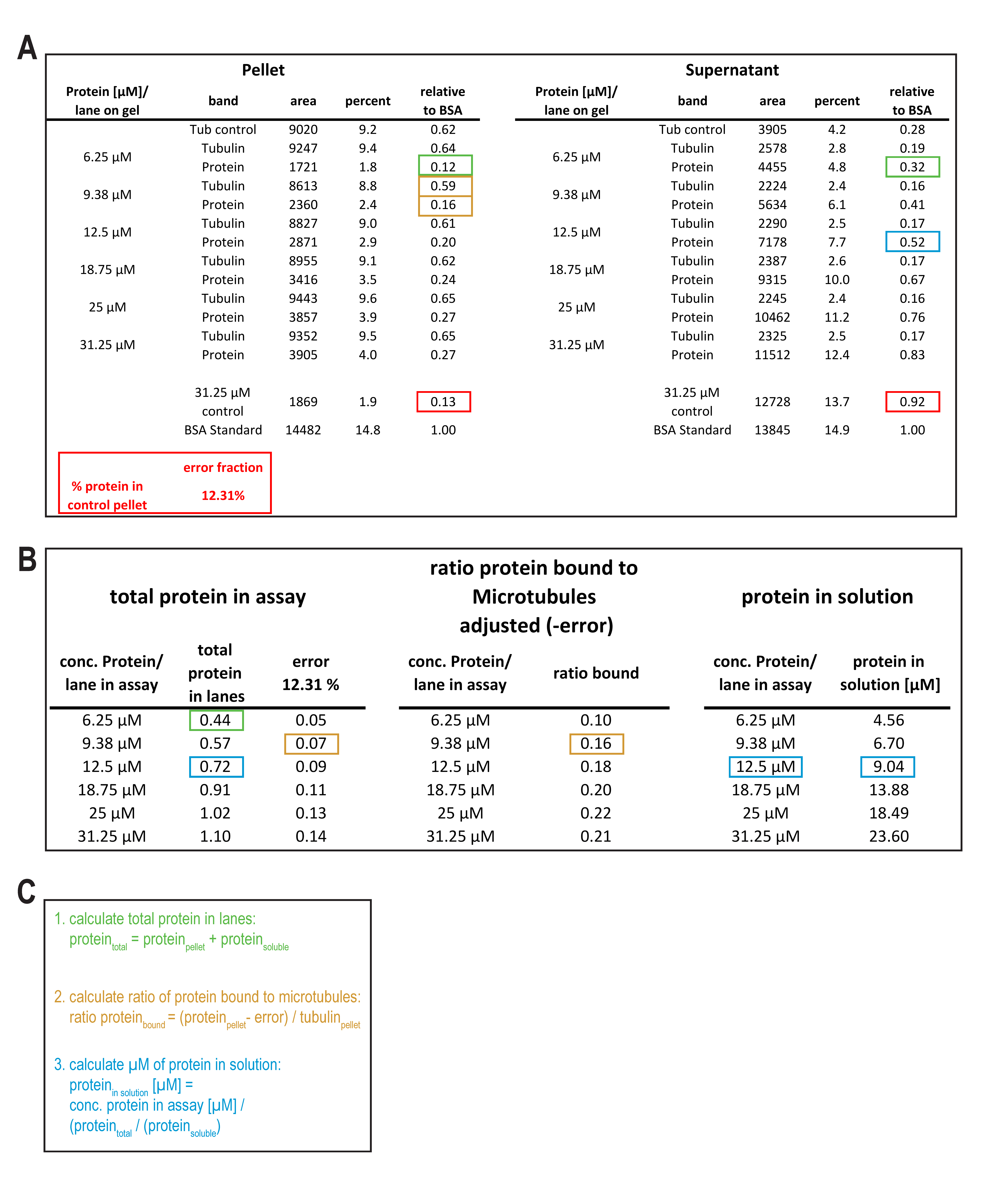
Figure 3. Representative data showing the calculations done on a data set obtained from 2 representative gels containing the pellet and supernatant fraction of 8 independent MT binding experiments as shown in Figure 2A. A. Calculation of relative peak areas in relation to the BSA standard on each gel. In case the protein of interest is also found in the pellet in absence of MTs, we recommend calculating the error fraction by normalizing the amount of protein found in the pellet by the total amount of protein in the experiment (see red boxes). B. Calculation of the total amount of protein, the ratio of protein bound to MTs and the amount of protein in solution. C. Equations used to calculate the values in B. Small boxes in A and B indicate exemplary values used in the equations. Color-coding: Equation 1-green, Equation 2-orange, Equation 3-blue. - Open GraphPad Prism and create a new XY project and choose the “Binding > Saturation binding, specific binding only” model.
- Copy the values of “% bound protein to microtubules” into the Y-column (see Figure 4A).
- Copy the values of “protein in solution (µM)” into the X-column (see Figure 4A).
- Press “Analyze” (highlighted in Figure 4A).
- Choose “Nonlinear regression (curve fit)”.
- The parameter options will open. Choose “Binding > Saturation”, “One site > Specific binding”.
- You will find your Kd and other statistical data (R2, confidence intervals etc.) in the “Results” tab.
- In the “Graphs” tab you find a graph displaying the fitted curve and your data points. Figure 4B shows a graph of the representative data used in this protocol.
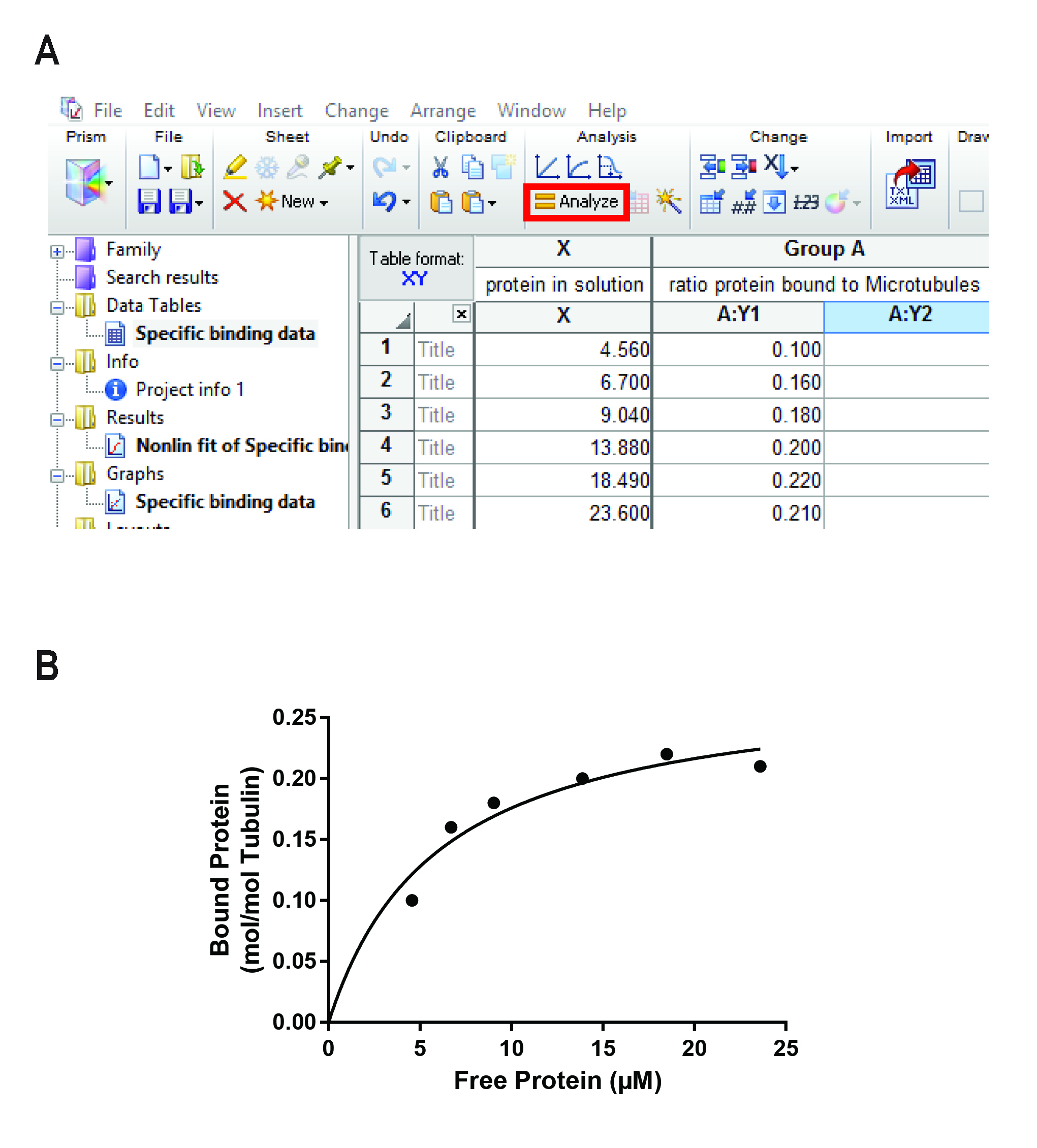
Figure 4. Analysis performed in GraphPad Prism. A. Analysis window of GraphPad Prism showing the data as calculated in Figure 3B. The “Analysis” button is highlighted. B. Representative curve obtained after fitting a saturation binding curve (see box) to the data shown in Figure 3B and Figure 4A.
Notes
- If the test protein is not stable in BRB80 buffer, one can exchange BRB80 to other buffers as long as taxol remains in the buffer to stabilize the MTs. An example of a representative experiment is shown in Figure 5. The same test protein as shown before was used but we replaced BRB80-T with 50 mM TRIS-HCl (pH 6.9), 100 µM taxol to stabilize MTs after polymerization (see section A, step 4). Under these conditions, no protein is detected in the pellet fraction in absence of MTs (lane 1). On the other hand, an increased abundance of tubulin in the supernatant is visible but this is typically not affecting the outcome of the assay as long as the amount of tubulin found in the supernatant is constant across all experiments.
- Keep in mind that high salt concentrations (> 50 mM, see above) can interfere with the binding of the test protein to MTs. High calcium concentrations (>1 mM) (O'Brien et al., 1997) were shown to depolymerize MTs. The pH of the buffer should be in a physiological range of approx. 6.0-8.5.
- If the test protein has the same molecular weight as tubulin (approx. 50 kDa) and they can thus not be separated by SDS-PAGE, it is possible to detect the protein and tubulin specifically in a western blot. In this case, we recommend using an antibody against the purification tag of the test protein and a specific antibody against α- or β-tubulin. The data analysis shown here is also working for membranes obtained via Western blotting.
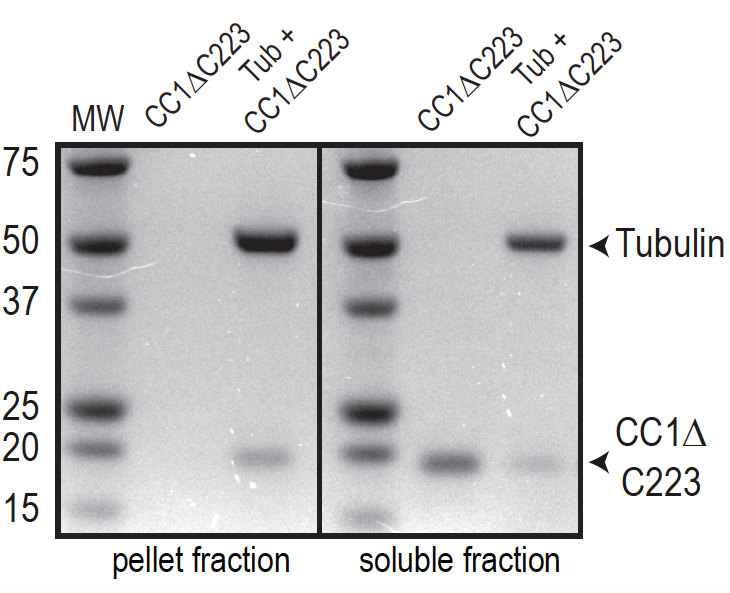
Figure 5. Representative data showing a MT binding assay. The test protein is the His-tagged, cytosolic domain of CC1 (CC1∆C223). In comparison to Figure 1A, BRB80-T was exchanged to 50 mM TRIS-HCl (pH 6.9), 100 µM taxol. Note that no protein is found in the pellet in absence of MTs because of enhanced protein stability. Vertical black lines denote spacing between two different gels.
Recipes
- Brinkley buffer 1980 (BRB80)
80 mM PIPES, adjusted to pH 6.9 using KOH
2 mM MgCl2
0.5 mM EGTA - BRB80 Cushion buffer
80 mM PIPES, adjusted to pH 6.9 using KOH
1 mM MgCl2
1 mM EGTA
60% glycerol - Taxol stocks
10 mM Taxol in DMSO
Split into 10 µl aliquots
Stored at -20 °C - GTP stocks
20 mM in BRB80
Split into 12.5 µl aliquots
Stored at -20 °C - Tubulin stocks
Keep on ice (!)
Mix 247.5 µl ice cold BRB80 with 12.5 µl GTP stock (= 250 µl BRB80 + 1 mM GTP)
Reconstitute 1 mg Tubulin with 250 µl ice cold BRB80 + 1 mM GTP (concentration = 4 mg/ml)
Gently flick the tube until tubulin is dissolved
Split into 10 µl aliquots on ice
Snap freeze in liquid nitrogen
Stored at -80 °C - MAP fraction stocks
Reconstitute 100 µg of the MAP fraction in 100 µl ice cold Milli-Q water (final MAP concentration of 1 mg/ml)
Split into 11 µl aliquots on ice
Snap freeze in liquid nitrogen
Stored at -80 °C - BSA stocks
Prepare a BSA solution in ice cold BRB80 with a concentration of 2.5 mg/ml
Split into 5 µl aliquots on ice
Snap freeze in liquid nitrogen
Stored at -80 °C - Colloidal Coomassie stain
Please follow the protocol of Dyballa and Metzger (2009) - Laemmli buffer
60 mM Tris-HCl (pH 6.8)
2% SDS
10% glycerol
5% β-mercaptoethanol
0.01% bromophenol blue
Acknowledgments
This protocol was adapted and modified from Goode et al. (1994), Gustke et al. (1994) and the datasheet from “Microtubule Binding Protein Spin-down Assay Kit” sold by Cytoskeleton, Inc. (http://www.cytoskeleton.com/pdf-storage/datasheets/bk029.pdf).
CK was funded from an IMPRS fellowship via the Max Planck Society and a Melbourne International Research Scholarship via the University of Melbourne. SP was supported by an R@MAP Professor position at UoM. Part of the research was funded through the DFG grant PE1642/6-1 and the ARC grant DP150103495.
References
- Dyballa, N. and Metzger, S. (2009). Fast and sensitive colloidal coomassie G-250 staining for proteins in polyacrylamide gels. J Vis Exp (30).
- Endler, A., Kesten, C., Schneider, R., Zhang, Y., Ivakov, A., Froehlich, A., Funke, N. and Persson, S. (2015). A mechanism for sustained cellulose synthesis during salt stress. Cell 162(6): 1353-1364.
- Goode, B. L. and Feinstein, S. C. (1994). Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter-repeat region of tau. J Cell Biol 124(5): 769-782.
- Gustke, N., Trinczek, B., Biernat, J., Mandelkow, E. M. and Mandelkow, E. (1994). Domains of tau protein and interactions with microtubules. Biochemistry 33(32): 9511-9522.
- Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259): 680-685.
- Mandelkow, E. and Mandelkow, E. M. (1995). Microtubules and microtubule-associated proteins. Curr Opin Cell Biol 7(1): 72-81.
- O’Brien, E. T., Salmon, E. D. and Erickson, H. P. (1997). How calcium causes microtubule depolymerization. Cell Motility and the Cytoskeleton 36(2): 125-135.
- Rosano, G. L. and Ceccarelli, E. A. (2014). Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol 5: 172.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Schindelin, J., Rueden, C. T., Hiner, M. C. and Eliceiri, K. W. (2015). The ImageJ ecosystem: An open platform for biomedical image analysis. Mol Reprod Dev 82(7-8): 518-529.
- Sivashanmugam, A., Murray, V., Cui, C., Zhang, Y., Wang, J. and Li, Q. (2009). Practical protocols for production of very high yields of recombinant proteins using Escherichia coli. Protein Sci 18(5): 936-948.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Kesten, C., Schneider, R. and Persson, S. (2016). In vitro Microtubule Binding Assay and Dissociation Constant Estimation. Bio-protocol 6(6): e1759. DOI: 10.21769/BioProtoc.1759.
Category
Plant Science > Plant cell biology > Cell structure
Plant Science > Plant developmental biology > General
Cell Biology > Cell structure > Cell adhesion
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.