- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

3’ Rapid Amplification of cDNA Ends (3’ RACE) Using Arabidopsis Samples





Published: Vol 5, Iss 19, Oct 5, 2015 DOI: 10.21769/BioProtoc.1604 Views: 21348
Reviewed by: Arsalan DaudiFang XuAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2015

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Production of functional eukaryotic RNA is a very elaborate process that involves a complex interplay between transcription and various RNA processing activities, including splicing, 5’ capping, and 3’ cleavage and polyadenylation (Bentley, 2014). Accurate mapping of RNA ends provides a valuable tool to assess transcriptional and post-transcriptional events giving rise to different gene transcripts. The abundance of such transcripts most likely depends on exogenous and developmental cues, or mutations. In the reference plant Arabidopsis, perturbation of the HUA-PEP post-transcriptional regulatory factors (Rodríguez-Cazorla et al., 2015) leads to the accumulation of aberrant transcripts of the key floral homeotic gene AGAMOUS (AG) (Yanofsky et al., 1990) that retain intronic sequence. It was determined by 3’ RACE reactions that such erroneous transcripts correspond to premature processing and polyadenylation events taking place at the AG intron region. Here we describe a protocol that is suitable for analysis of relatively abundant transcripts and also for detecting aberrant RNA species that are likely prone to rapid turnover. Likewise, the method, here adapted to Arabidopsis reproductive tissues, can be applied to characterize RNA species from other organs (leaf, root) and/or other plant species. We provide a detailed protocol of our 3’ RACE procedure comprising four major parts: Total RNA extraction, RNA amount determination and quality control, the RACE procedure itself, and isolation of the resulting RACE products for cloning and sequencing.
Keywords: ArabidopsisMaterials and Reagents
- Disposable gloves
- Sterile disposable RNase-free pipette tips
- RNase-free microcentrifuge tubes
- Plant sample (Arabidopsis young flower buds, stages 1 through 9)
Note: Arabidopsis flower stages according to Smyth et al., 1990. Other tissue/species can be tested as well. - Liquid nitrogen
- GeneJET Plant RNA Purification Kit (Thermo Fisher Scientific, catalog number: K0801 )
- 1 M DTT (Sigma-Aldrich, catalog number: 43816 )
- Absolute ethanol (JT Baker 8006) and 96% Ethanol (as recommended by the RNA extraction kit manufacturer, see point 6 above)
- 4M LiCl (made in distilled water and autoclaved, not necessarily fresh)
- DNase I, RNase-free (Thermo Fisher Scientific, catalog number: EN0525 )
- RiboLock RNase Inhibitor (Thermo Fisher Scientific, catalog number: EO0382 )
- dNTPs mix 10 mM each (Thermo Fisher Scientific, catalog number: R0192 )
- OligodT-Anchor Primer (Roche 5’/3’ RACE Kit) (Roche Diagnostics, catalog number: 03 353 621 001 , version 10)
- Maxima Reverse Transcriptase + buffer 5x (Thermo Fisher Scientific, catalog number: EP0741 )
- RNase-free water
- High Fidelity PCR Enzyme Mix + buffer 10x (Thermo Fisher Scientific, catalog number: K0191 )
- PCR Anchor Primer (Roche 5’/3’ RACE Kit) (Roche Diagnostics, catalog number: 03 353 621 001 )
- Gene specific 5’ Primer/s (Table 1)
- GeneJET Gel Extraction Kit (Thermo Fisher Scientific, catalog number: K0691 )
- StrataClone PCR Cloning Kit (Agilent Technologies, catalog number: 240205 )
- Taq DNA Polymerase (EURx, catalog number: EK2500-04 )
- GeneRulerTM 100 bp Plus DNA Ladder (Thermo Fisher Scientific, catalog number: SM0321 ) (or any other suitable molecular marker for your convenience)
Equipment
- Mortar and pestle (beaked Haldenwanger mortar, 63 mm. diameter. Clean, autoclave and dry before use)
- Thermomixer (water bath or heat block can be used as well) (Eppendorf)
- Microcentrifuge (Heraeus, Biofuge Pico)
- Refrigerated centrifuge BR15 (B. Braun Melsungen AG) with rotor 12,148-H (Sigma-Aldrich) for 1.5 ml Eppendorf tubes
- BioPhotometer Plus (Eppendorf)
- Thermal Cycler T100 (Bio Rad) (or any other conventional PCR device)
- Electrophoresis system
- UV transilluminator
Procedure
- Extracting total RNA from flower buds
- Pour liquid nitrogen in the mortar, let it to evaporate and then carefully place plant material. Grind until a fine homogeneous powder is obtained. Store in a 1.5 ml Eppendorf tube with a small spatula.
Note: Operate at room temperature. Plant material can be freshly collected or previously harvested and stored at -80 °C. In both cases, however, harvesting must be carried out under freezing conditions (it is recommended to keep the Eppendorf tube and spatula in liquid nitrogen prior to use). - Extract total RNA by using GeneJET Plant RNA Purification Kit (see protocol).
- Add 20 μl of 1 M DTT to each 500 μl of RNA Lysis Solution (containing the chaotropic salt guanidine thiocyanate). Add this mixture to the tube containing the tissue powder (up to 100 mg) and vortex for 10-20 sec to mix thoroughly. This step should be performed quickly to avoid degradation.
- Incubate for 3 min at 56 °C in a thermomixer or heat block.
- Centrifuge for 5 min at maximum speed (≥20,000 x g, ≥13,000-14,000 rpm).
- Collect the supernatant (usually 450-550 μl) and transfer to a clean 1.5 ml tube.
- Add 250 μl of 96% ethanol. Mix by pipetting.
- Transfer the mixture to a purification column inserted in a 2 ml collection tube (both provided with the kit).
- Centrifuge the column for 1 min at 12,000 x g (~11,000 rpm). Discard the flow-through solution and reassemble column and collection tube.
- Add 700 μl of Wash Buffer WB 1 (provided with the kit) to the purification column.
- Centrifuge for 1 min at 12,000 x g (~11,000 rpm). Discard the flow-through and collection tube. Place the purification column into a clean 2 mL collection tube.
- Add 500 μl of Wash Buffer 2 (provided with the kit) to the purification column.
- Centrifuge for 1 min at 12,000 x g (~11,000 rpm). Discard the flow-through solution and reassemble column and collection tube.
- Repeat previous steps A2j-k and re-spin the column for 1 min at maximum speed to remove all traces of Wash Buffer 2.
- Elute the RNA with 50 μl of nuclease-free water and centrifuge for 1 min at 12,000 x g (~11,000 rpm). Repeat this step with the same volume of nuclease-free water to increase the yield of RNA.
- Use the purified RNA immediately or store until use. Storage at -80 °C is recommended.
- Add 20 μl of 1 M DTT to each 500 μl of RNA Lysis Solution (containing the chaotropic salt guanidine thiocyanate). Add this mixture to the tube containing the tissue powder (up to 100 mg) and vortex for 10-20 sec to mix thoroughly. This step should be performed quickly to avoid degradation.
- DNase treatment and concentration.
- Treat the sample with 5 µl (5 U) of DNase I (Thermo Fisher Scientific, according to manufacturer’s protocol with minor modifications) for 1 h at 37 °C to eliminate traces of genomic DNA. Include RiboLock RNase Inhibitor at 1 U/µl to prevent RNA degradation.
- Add 10 µl 50 mM EDTA and incubate at 65 °C for 10 min.
- Immediately, mix the RNA sample with two volumes of cold 100% ethanol and store at -20 °C for several hours or overnight.
- Add 1/10 volume of 4 M LiCl and centrifuge at 12,000 rpm and 4 °C for 10 min. Remove the supernatant carefully.
- Wash with 500 µl cold 70% ethanol and centrifuge as in step A3d.
- Remove as much supernatant as possible with the pipette tip. Put the microcentrifuge tube with the open lid in a box with ice, and let the pellet dry for 5 min at the bench or in a laminar flux cabin.
Note: Drying the pellet at room temperature also works fine. - Resuspend in 25 μl RNase-free water and store at -80 °C if not used immediately.
- Determine RNA concentration photometrically, and check integrity by visualizing 500 ng in a 2% agarose gel (Figure 1).
Note: It is highly recommended to use photometer cuvettes and gel trays previously treated with 0.4 M NaOH (at least for 5 min) to minimize the presence of RNases. - Control PCR. Despite DNase I treatment after RNA extraction, 0.5 µl of the sample was used as template for a conventional PCR reaction with deoxynucleotide primers to test absence of genomic DNA. Carry out PCR with a pair of primers of your choice to test the presence of DNA in your sample. Actin or other housekeeping genes might be suitable.
PCR components:
16 µl Distilled water
2.5 µl 10x Buffer B (+15 mM MgCl2)
1 µl dNTPs mix (5 mM each)
1 µl ACT2-f (5 µM)(see oligonucleotide sequence below)
1 µl ACT2-r (5 µM)(see oligonucleotide sequence below)
0.25 µl (1.25 U) Taq DNA polymerase
0.5 µl RNA simple
PCR profile:1 cycle 94 °C, 2 min 35 cycles 94 °C, 30 sec; 51 °C, 30 sec; 72 °C, 30 sec 1 cycle 72 °C, 10 min
- Treat the sample with 5 µl (5 U) of DNase I (Thermo Fisher Scientific, according to manufacturer’s protocol with minor modifications) for 1 h at 37 °C to eliminate traces of genomic DNA. Include RiboLock RNase Inhibitor at 1 U/µl to prevent RNA degradation.
- Pour liquid nitrogen in the mortar, let it to evaporate and then carefully place plant material. Grind until a fine homogeneous powder is obtained. Store in a 1.5 ml Eppendorf tube with a small spatula.
- 3’ RACE
- Designing specific primers (forward primers).
Primers were designed using the free online software OligoCalc (http://www.basic.northwestern.edu/biotools/oligocalc.html). Best suited primers usually are 20 to 25 nt in length, and 50-60% GC content (as suggested by the Roche 5’/3’ RACE Kit protocol). Melting temperature (Tm) must be close to that of the PCR Anchor primer (see below), not exceeding a 5 °C difference. - Retrotranscription.
According to the instructions provided by the Maxima Reverse Transcriptase protocol with minor modifications.- Mix the following components for the annealing reaction mixture:
5 µg of previously isolated total RNA
1 µl of Oligod(T)-Anchor Primer (37.5 µM) from Roche 5’/3’ RACE Kit (A polydT stretch followed by an adapter sequence, see primer sequence below)
1 µl dNTPs mix (10 mM each)
14.5 µl Distilled water (nuclease-free) - Incubate at 65 °C for 5 min. Chill on ice, centrifuge for a few seconds (to place the whole reaction at the bottom) and place on ice again.
- Add the following reaction components to the annealing mixture in the indicated order
4 µl 5x RT Buffer (provided with the enzyme batch)
0.5 µl (20 U) RiboLock
1 µl (200 U) Maxima Reverse Transcriptase - Incubate at 60 °C for 30 min.
Note: Retrotranscription of GC-rich RNAs can be performed at 65 °C. Indeed, temperature may vary from 50 to 65 °C. Pilot experiments might be necessary to find optimal temperature. - Terminate the reaction by heating at 85 °C for 5 min.
- Mix the following components for the annealing reaction mixture:
- Retrotranscription verification.
This is an optional step. Carry out a conventional PCR procedure with a pair of primers of your choice to corroborate the presence of cDNA in your sample. Actin or other housekeeping genes might be suitable. - First PCR (PCR amplification of cDNA). According to the High Fidelity PCR Enzyme Mix protocol with some modifications.
- Prepare the following reaction mixture
30 µl Distilled water
5 µl 10x High Fidelity PCR buffer (+ 15 mM MgCl2)
1 µl dNTPs mix, 10 mM each
1 µl specific primer (forward primer), 12.5 µM
5 µl Template DNA
0.4 µl (2 U) High Fidelity PCR Enzyme Mix
8 µl PCR Anchor Primer (reverse primer) 12.5 µM (from Roche 5’/3’ RACE Kit, corresponding to the adapter sequence. See primer sequence below) - PCR profile (check manufacturer’s indications, parameters may vary according to the length of the expected products)
1 cycle 94 °C, 2 min
35 cycles 94 °C, 30 sec; 57 °C, 30 sec; 68 °C, 4 min 30 sec
1 cycle 68 °C, 10 min
Note: It might be convenient to perform simultaneous reactions with distinct forward primers (2-4) hybridizing at close locations in order to select that one yielding optimal results. - Check 10 µl of the reaction in an agarose gel (Figure 2)
Note: Agarose concentration and running time may depend on the size of the expected products.
- Prepare the following reaction mixture
- Second PCR. Same as in step B4, except template (an aliquot of the first strand PCR), and a new forward primer (hybridizing downstream to the one used in the first reaction to endorse specificity).
- Reaction mixture
34 µl Distilled water
5 µl 10x High Fidelity PCR buffer (+ 15 mM MgCl2)
1 µl dNTPs mix, 10 mM each
1 µl specific primer (forward primer), 12.5 µM
8 µl PCR Anchor Primer (reverse primer) 12.5 µM
1 µl First PCR reaction
0.4 µl (2 U) High Fidelity PCR Enzyme Mix - Use same PCR profile as in step B4b.
Note: Eventually the forward primer might be one of those tested for the first strand reaction provided that it lies downstream to the one used during that step.
- Reaction mixture
- Isolate PCR products.
- Load the whole reaction in an agarose gel and carry out electrophoresis until the bands are separated as much as possible (Figure 3).
Note: Agarose concentration may depend on the length of the expected products. - Under a UV transilluminator, excise PCR DNA bands and store separately in Eppendorf tubes.
- Purify DNA using GeneJET Gel Extraction Kit (see protocol) or any other equivalent system. Finally, DNA is eluted from columns in a total volume of 30 µl.
- Load the whole reaction in an agarose gel and carry out electrophoresis until the bands are separated as much as possible (Figure 3).
- Cloning and sequencing.
Eluted dsDNA PCR fragments contain 3'-A overhangs that allow for easy cloning in any T-vector plasmid system. For example, see protocol for StrataClone PCR Cloning Kit. Finally, sequence and analyze the structure of inserts contained in positive clones.
- Designing specific primers (forward primers).
Representative data
Table 1. Oligonucleotides used in this study
| Name | Sequence (5’ – 3’) | References |
| Oligo d(T)-Anchor Primer | GACCACGCGTATCGATGTCGACTTTTTTTTTTTTTTTTV | Roche 5’/3’ RACE Kit |
| PCR Anchor Primer | GACCACGCGTATCGATGTCGACRoche | 5’/3’ RACE Kit |
| ACT2-f | CTCTTAACCGTAAAGCTAACAG | An et al., 1996 |
| ACT2-r | AGTGAGAATCTTCATGAGTGAG | An et al., 1996 |
| AGIa (specific forward primer) | CGGATCGAGAACACAACGAATCG | Rodríguez-Cazorla et al., 2015 |
| AGIb (specific forward primer) | GGTTTGCTCAAGAAAGCTTACGAGC | Rodríguez-Cazorla et al., 2015 |
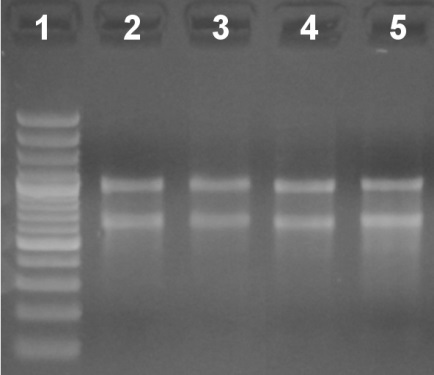
Figure 1. Relative quantitation and qualitative analysis of total RNA in a 2% agarose gel stained with ethidium bromide (EtBr). Lane 1: Molecular weight ladder. Lanes 2-5: 500 ng of total RNA from different samples.
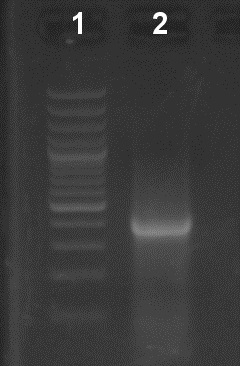
Figure 2. Verification of the first PCR step in an EtBr-stained agarose gel (1%). Lane 1: Molecular weight ladder. Lane 2: 10 µl of the first PCR reaction.
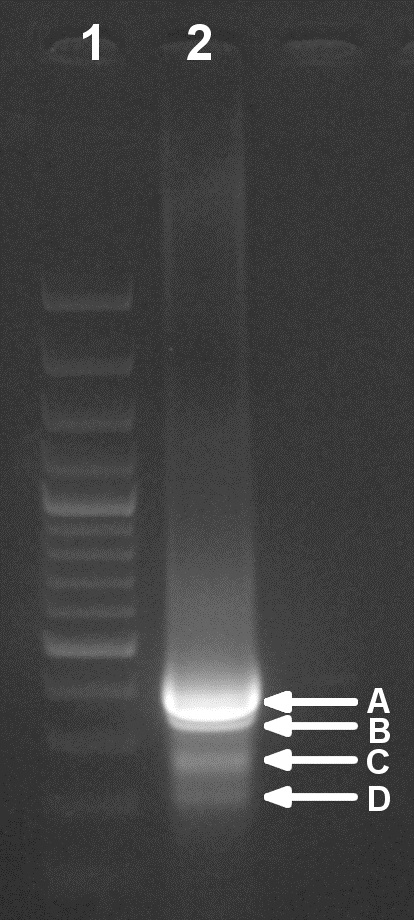
Figure 3. Verification of the second PCR step in an EtBr-stained agarose gel (1.5%). Lane 1: Molecular weight ladder. Lane 2: Total PCR sample (50 µl). Four bands of different size can be distinguished (A to D) that could be excised and purified as indicated in steps B6b-c.

Figure 4. Schematic representation of examples of prematurely processed AG transcripts identified by RACE. DNA sequence corresponding to exon 2 appears as white upper-case letters boxed in black. Intron 2 sequence is shown as lower-case black letters. Cleavage site is indicated (A in red). Additional representative data could also refer to Figures 4 and S11 from Rodríguez-Cazorla et al. (2015).
Acknowledgments
This work was supported by grants from Ministerio de Economía y Competitividad of Spain (https://sede.micinn.gob.es) (grant BIO2014-56321-P to AV) and National Science Foundation of USA (http://www.nsf.gov/; grant IOS-1121055 to MFY) and Paul D. Saltman Endowed Chair in Science Education (http://biology.ucsd.edu/news/awards-and-honors/endowedchairs.html) to MFY.
References
- An, Y. Q., McDowell, J. M., Huang, S., McKinney, E. C., Chambliss, S. and Meagher, R. B. (1996). Strong, constitutive expression of the Arabidopsis ACT2/ACT8 actin subclass in vegetative tissues. Plant J 10(1): 107-121.
- Bentley, D. L. (2014). Coupling mRNA processing with transcription in time and space. Nat Rev Genet 15(3): 163-175.
- Nojima, T., Gomes, T., Grosso, A. R., Kimura, H., Dye, M. J., Dhir, S., Carmo-Fonseca, M. and Proudfoot, N. J. (2015). Mammalian NET-Seq reveals genome-wide nascent transcription coupled to RNA processing. Cell 161(3): 526-540.
- Rodriguez-Cazorla, E., Ripoll, J. J., Andujar, A., Bailey, L. J., Martinez-Laborda, A., Yanofsky, M. F. and Vera, A. (2015). K-homology nuclear ribonucleoproteins regulate floral organ identity and determinacy in arabidopsis. PLoS Genet 11(2): e1004983.
- Smyth, D. R., Bowman, J. L. and Meyerowitz, E. M. (1990). Early flower development in Arabidopsis. Plant Cell 2(8): 755-767.
- Yanofsky, M. F., Ma, H., Bowman, J. L., Drews, G. N., Feldmann, K. A. and Meyerowitz, E. M. (1990). The protein encoded by the Arabidopsis homeotic gene agamous resembles transcription factors. Nature 346(6279): 35-39.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Rodríguez-Cazorla, E., Andújar, A., Ripoll, J. J., Bailey, L. J., Martínez-Laborda, A., Yanofsky, M. F. and Vera, A. (2015). 3’ Rapid Amplification of cDNA Ends (3’ RACE) Using Arabidopsis Samples. Bio-protocol 5(19): e1604. DOI: 10.21769/BioProtoc.1604.
Category
Plant Science > Plant molecular biology > DNA > Gene expression
Plant Science > Plant molecular biology > RNA > Transcription
Molecular Biology > DNA > PCR
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.