- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Pyridine Hemochromagen Assay for Determining the Concentration of Heme in Purified Protein Solutions


Published: Vol 5, Iss 18, Sep 20, 2015 DOI: 10.21769/BioProtoc.1594 Views: 33587
Reviewed by: Fanglian HeSeda Ekici
Original research article
The authors used this protocol in:

Jul 2012
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Heme is a common cofactor in proteins, found in hemoglobin, myoglobin, cytochrome P450, DGCR8, and nitric oxide synthase, among others. This protocol describes a method for quantifying heme that works best in purified protein samples. This protocol might be used to, for example, determine whether a given heme-binding protein is fully occupied by heme, thus allowing correlation of heme content with activity. This requires the absolute heme concentration and an accurate protein concentration. Another use is to determine the extinction coefficients of a heme-bound protein. This assay is fast, easy, and reproducible if done correctly.
Keywords: hemeBackground
G.G. Stokes was the first to prepare what we now refer to as hemochromagen. As early as 1863, he was monitoring changes in the hemoglobin absorbance spectrum upon reduction of the heme to the Fe(II) form Stokes (1863). Stokes had reduced blood in the presence of ammonia; what he was seeing were the intense α and β peaks of Fe(II) heme b from hemoglobin in complex with ammonia. Later authors (Anson and Mirsky, 1928) were able to show that the hemochromagen, as it had been called by Christian Bohr (Edsall, 1972) , was heme in complex with some nitrogenous ligand. It can be formed as well by simply reducing myoglobin under denaturing conditions, in which case histidines serve as the axial ligands. Hill demonstrated that the pyridine hemochromagen is formed from the nitrogen of two pyridine molecules binding to the axial position of the reduced heme (Hill, 1926; Hill, 1929); this was confirmed by Smith (1959) and Gallagher and Elliot (Gallagher et al., 1965; Gallagher et al., 1968).
With advances in spectroscopic techniques, later workers were able to use hemochromagen for an important analytical purpose (Hill, 1929). The regularity of the α peak of reduced pyridine hemochromagen, its high extinction coefficient, and the fact that it follows Beer's Law over a wide range allows its use for determining the total heme composition of a sample. De Duve (1948) published one of the first protocols for this, and determined the extinction coefficient (ε) at 557 nm to be 32 mM-1 cm-1. The method used relied on gravimetric determination of the standard heme samples, which could lead to an underestimate of the true heme concentration if the sample is impure. Paul et al. (1953) re-determined the extinction coefficient for pyridine hemochromogen from recrystallized heme b and myoglobin, using the iron content as an internal control. They found a value (34.7 mM-1 cm-1) significantly higher than the previous value of 32 mM-1 cm-1, representing a difference of roughly 9%. Both values have been in use comparatively recently: e.g. (Scott and Lecomte, 2000; Fushitani and Riggs, 1988; Miyoshi et al., 1997; Yachie et al., 1999; Lee et al., 2000; Huche et al., 2006), which use 32 mM-1 cm-1, and (Sono et al., 1984; Senturia et al., 2012; Sinclair et al., 2001; Berry and Trumpower, 1987), which use 34.7 mM-1 cm-1. In this assay we have taken the value given by Paul et al. (1953) to be most accurate.
[Principle of action] The heme-containing protein solution is first mixed with Solution I (see Recipes) containing pyridine, NaOH and potassium ferricyanide. Pyridine serves as a ligand for heme in the Fe(II) state. NaOH keeps sodium dithionite stable. Potassium ferricyanide ensures that all dissolved heme is in the Fe(III) state at first. Then, excess sodium dithionite in solution is added to reduce the heme from Fe(III) to Fe(II). Another option is to use a pipette tip to add a few grains of solid dithionite to the cuvette, and allow it to dissolve. Recipes similar to the current one have been recommended by Sinclair et al. (2001); Berry and Trumpower (1987); Antonini and Brunori (1971). The concentration of heme in the sample can be determined from the absorbance of the reduced sample; the extinction coefficient for reduced pyridine hemochromagen is 34.7 mM-1 cm-1 at 557 nm for heme b. It can also be done using the difference spectrum between the reduced and oxidized samples. Don't forget, in either case, to take your dilution factor into consideration. This protocol is written for heme b; however, other types of heme form hemochromagens as well and can be quantified using the same technique. See Berry and Trumpower (1987) or Table 1 for extinction coefficients for hemes a and c.
Materials and Reagents
- Heme-containing sample
Note: The concentration of heme in your stock solution should be at least 10 μM and not much greater than 80 μM. Dithiothreitol (DTT) and other reducing agents can reduce potassium ferricyanide and may interfere with the oxidized sample, but not with the reduced sample. Any common biological buffer should be compatible with this procedure so long as it has no significant absorbance in the 500 nm to 600 nm range and does not form a complex with heme, as should be the case with all Good's buffers. We have personally used phosphate, tris, HEPES, EPPS, MES, and CHES without issue. - 0.5 M NaOH
- Pyridine (Sigma-Aldrich, catalog number: 360570 )
- Potassium ferricyanide(III) (Sigma-Aldrich, catalog number: 702587 )
- Sodium dithionite (Sigma-Aldrich, catalog number: 157953-5 G )
- Deionized water
- 0.2 M NaOH, 40% (v/v) pyridine, 500 μM potassium ferricyanide (see Recipes)
- 0.1 M potassium ferricyanide (K3[Fe(CN)6]) (see Recipes)
- 0.5 M sodium dithionite in 0.5 M NaOH (see Recipes)
Equipment
- Spectrophotometer with a bandwidth ≤ 2 nm
- Fume hood
- Quartz or glass cuvette, 1 cm length
- Pipettes
Note: It is generally good to have a spectrophotometer capable of relatively low spectral bandwidth (SBW) in order to get highly accurate measurements. It is suggested in the literature that the ratio of SBW to the natural bandwidth (NBW) should be 0.1 or lower in order to get an error of less than 0.5% (Surles and Erickson, 1974; Brodersen, 1954). The α band of reduced pyridine hemochromagen has a NBW of around 20 nm, hence the recommendation of 2 nm or less for SBW. Check with your manufacturer if you are unsure of your SBW. Most modern spectrophotometers have SBW less than 4 nm, corresponding to an error of less than 2%. It is also important to note that this error is not random, but results in an underestimate of the true absorbance; having a higher SBW/NBW ratio leads to a 'flattening' of absorbance peaks. If possible, use a scanning spectrophotometer. This allows you to verify that your spectrum looks similar to the spectrum shown in Figure 1.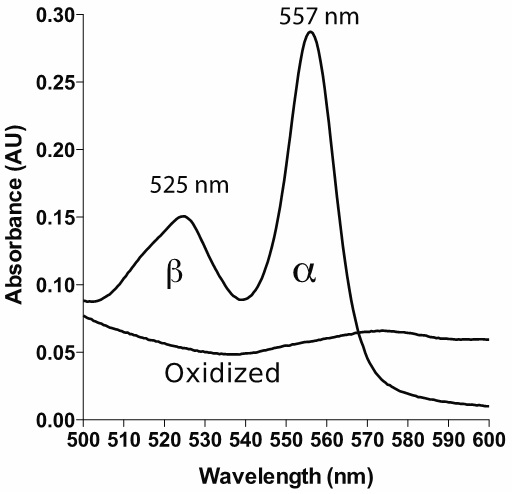
Figure 1. Example spectrum of reduced and oxidized pyridine hemochromagen (heme b). The bandwidth is set to 1 nm, with data interval 1 nm.
Procedure
The volumes shown below can be scaled up or down as desired, as long as the concentrations stay the same.
- Set the spectrophotometer to ≤ 2 nm SBW (if adjustable) and data interval at 1 nm between 500 and 600 nm (at least). The assay is not strongly temperature sensitive; room temperature works well.
- Blank the spectrophotometer with Solution I mixed 1:1 with whichever buffer your protein sample is in.
- Empty cuvette. Add 0.5 ml Solution I to 0.5 ml of your heme-containing sample and mix well.
- Scan this mixture; this is your oxidized sample.
- Add 10 μl Solution III to your oxidized sample and mix well. The sample should turn a reddish color.
- Scan immediately and again every minute until the absorbance peak no longer increases; this should take no more than 5 min. The scan with the highest peak is the reduced spectrum.
- Using the extinction coefficients in Table 1, and taking into account your dilution factors, calculate the concentration of heme in your original sample using Beer's law, A = ε c l (Absorbance = extinction coefficient x concentration x pathlength). You can use the absolute absorbance of the reduced sample, or the difference spectrum.
Table 1. Extinction coefficients for pyridine hemochromogensCompound Wavelength ε (mM-1 cm-1) Solvent Ref. Pyr2-heme b Reduced 557 nm 34.7 NaOH, 10-40% pyr. Paul et al., 1953 Pyr2-heme b, Red. - Oxid. 557 nm min 540 nm 23.98 NaOH, 10-40% pyr. Berry and Trumpower, 1987 Pyr2-heme a, Red. - Oxid. 587 nm min 620 nm 23.98 NaOH, 10-40% pyr. Berry and Trumpower, 1987 Pyr2-heme c Reduced 550 nm 30.27 NaOH, 10-40% pyr. Berry and Trumpower, 1987 Pyr2-heme c, Red. - Oxid. 550 nm min 535 nm 23.97 NaOH, 10-40% pyr. Berry and Trumpower, 1987 - To determine the extinction coefficient of the heme when it is bound to your native protein, you will also need a high quality scan of the protein sample in a suitable buffer. Using Beer's law and the sample heme concentration, calculate ε for the heme absorbance peaks of your native protein.
Recipes
- Solution I [0.2 M NaOH, 40% (v/v) pyridine, 500 μM potassium ferricyanide]
Add 2/5 volume 0.5 M NaOH to 2/5 volume pyridine in fume hood or well-ventilated space and dilute to final volume with water
Add 1/200 volume 0.1 M potassium ferricyanide (Solution II)
Paul et al. (1953) reported that NaOH concentrations between 0.02 and 0.5 M and pyridine concentrations between 10% and 50% make no difference to the molar absorptivities of the hemochromagen.
Hazards: Pyridine is toxic, flammable, and has what the Merck Index calls a "characteristic disagreeable odor." Mix solutions in a fume hood or well-ventilated area.
Warnings: Pyridine eventually oxidizes; when this happens it turns yellow. Do not use if yellow, and seal the pyridine bottle after use. - Solution II {0.1 M potassium ferricyanide (K3[Fe(CN)6])}
Hazards: Releases cyanide if mixed with strong acids.
Warnings: Potassium ferricyanide must be made fresh and used within 24 h. Dispose of in a separate container. - Solution III (0.5 M sodium dithionite in 0.5 M NaOH)
Sodium dithionite, off the shelf, is usually no more than 85% pure, no matter the supplier (McKenna et al., 1991).
Note: It should always be dissolved and diluted in neutral or basic solution; acidic conditions cause it to release hydrogen sulfide, a poisonous gas. It must be made fresh every time.
Storage: Sodium dithionite should be kept away from water. Fresh sodium dithionite is powdery and free-flowing. It is best to purchase it in small amounts and open a fresh bottle periodically, due to degradation upon exposure to air (McKenna et al., 1991). Sodium dithionite has a redox potential of -0.66 V at pH 7.0, 25 °C (Mayhew, 1978), and doesn’t absorb strongly in the > 400 nm region.
Acknowledgments
This work was funded in part by a grant from the NIH (GM080563) to F. G. and a UCLA Dissertation Year Fellowship to I. B. We would also like to thank Aaron T. Smith and Judith N. Burstyn for advice.
References
- Anson, M. L. and Mirsky, A. E. (1928). On Hemochromogen. J Gen Physiol 12(2): 273-288.
- Antonini, E. and Brunori, M. (1971). Hemoglobin and myoglobin in their reactions with ligands. North-Holland Pub. Co.
- Berry, E. A. and Trumpower, B. L. (1987). Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra. Anal Biochem 161(1): 1-15.
- Brodersen, S. (1954). Slit-width effects. J Opt Soc Am 44:22.
- De Duve, C. (1948). A spectrophotometric method for the simultaneous determination of myoglobin and hemoglobin in extracts of human muscle. Acta Chem Scand 2(3): 264-289.
- Edsall, J. T. (1972). Blood and hemoglobin: the evolution of knowledge of functional adaptation in a biochemical system, part I: The adaptation of chemical structure to function in hemoglobin. J Hist Biol 5(2): 205-257.
- Fushitani, K. and Riggs, A. F. (1988). Non-heme protein in the giant extracellular hemoglobin of the earthworm Lumbricus terrestris. Proc Natl Acad Sci U S A 85(24): 9461-9463.
- Gallagher, W. A. and Elliott, W. B. (1965). The formation of pyridine haemochromogen. Biochem J 97(1): 187-193.
- Gallagher, W. A. and Elliott, W. B. (1968). Alkaline haematin and nitrogenous ligands. Biochem J 108(1): 131-136.
- Hill, R. (1926). The chemical nature of haemochromogen and its carbon monoxide compound. Proc R Soc London B Biol Sci 419-430.
- Hill, R. (1929). Reduced haematin and haemochromogen. Proc R Soc London Ser B Contain Pap a Biol 112-130.
- Huche, F., Delepelaire, P., Wandersman, C. and Welte, W. (2006). Purification, crystallization and preliminary X-ray analysis of the outer membrane complex HasA-HasR from Serratia marcescens. Acta Crystallogr Sect F Struct Biol Cryst Commun 62(Pt 1): 56-60.
- Lee, Y. C., Martin, E. and Murad, F. (2000). Human recombinant soluble guanylyl cyclase: expression, purification, and regulation. Proc Natl Acad Sci U S A 97(20): 10763-10768.
- Mayhew, S. G. (1978). The redox potential of dithionite and SO-2 from equilibrium reactions with flavodoxins, methyl viologen and hydrogen plus hydrogenase. Eur J Biochem 85(2): 535-547.
- McKenna, C. E., Gutheil, W. G. and Song, W. (1991). A method for preparing analytically pure sodium dithionite. Dithionite quality and observed nitrogenase-specific activities. Biochim Biophys Acta 1075(1): 109-117.
- Miyoshi, S., Inami, Y., Moriya, Y., Kamei, T., Rahman, M. M., Yamamoto, S., Tomochika, K. and Shinoda, S. (1997). Characterization of a mutant of Vibrio vulnificus for heme utilization. FEMS Microbiol Lett 148(1): 101-106.
- Paul, K. G., Theorell, H. and Åkeson, A. (1953). The molar light absorption of pyridine ferroprotoporphyrin (pyridine haemochromogen). Acta Chem Scand 7 1284-1287.
- Scott, N. L. and Lecomte, J. T. (2000). Cloning, expression, purification, and preliminary characterization of a putative hemoglobin from the cyanobacterium Synechocystis sp. PCC 6803. Protein Sci 9(3): 587-597.
- Senturia, R., Laganowsky, A., Barr, I., Scheidemantle, B. D. and Guo, F. (2012). Dimerization and heme binding are conserved in amphibian and starfish homologues of the microRNA processing protein DGCR8. PLoS One 7(7): e39688.
- Sinclair, P. R., Gorman, N. and Jacobs, J. M. (2001). Measurement of heme concentration. Curr Protoc Toxicol Chapter 8: Unit 8 3.
- Smith, M. H. (1959). Kinetics and equilibria in systems containing haem, carbon monoxide and pyridine. Biochem J 73: 90-101.
- Sono, M., Dawson, J. H. and Hager, L. P. (1984). The generation of a hyperporphyrin spectrum upon thiol binding to ferric chloroperoxidase. Further evidence of endogenous thiolate ligation to the ferric enzyme. J Biol Chem 259(21): 13209-13216.
- Stokes, G. G. (1863). On the reduction and oxidation of the colouring matter of the blood. Proc R Soc 13: 355-364.
- Surles, T. and Erickson, J. O. (1974). Absorbance measurements at various spectral bandwidths. Clin Chem 1243-1244.
- Yachie, A., Niida, Y., Wada, T., Igarashi, N., Kaneda, H., Toma, T., Ohta, K., Kasahara, Y. and Koizumi, S. (1999). Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest 103(1): 129-135.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Barr, I. and Guo, F. (2015). Pyridine Hemochromagen Assay for Determining the Concentration of Heme in Purified Protein Solutions. Bio-protocol 5(18): e1594. DOI: 10.21769/BioProtoc.1594.
Category
Biochemistry > Other compound > Heme
Biochemistry > Protein > Interaction > Protein-ligand interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.