- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

13C Kinetic Labeling and Extraction of Metabolites from Adherent Mammalian Cells
Published: Vol 5, Iss 8, Apr 20, 2015 DOI: 10.21769/BioProtoc.1447 Views: 11323
Reviewed by: Fanglian HeValentine V TrotterKanika Gera
Original research article
The authors used this protocol in:

Jul 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Fluctuations in metabolite levels in mammalian cells are the most direct form of readout of the cellular metabolic state. The current protocol describes a method for pulse labeling and subsequent isolation of metabolites from adherent mammalian cells. The isolated metabolites can be identified and quantified by mass-spectrometry, allowing for estimation of the rates of synthesis and removal of metabolites from the system being analyzed.
Materials and Reagents
- THP1 cells (from ATCC, catalog number: TIB 202 )
- Methanol (LC-MS Ultra CHROMASOLV) (Sigma-Aldrich, catalog number: 14262 )
- Water (LC-MS Ultra CHROMASOLV) (Sigma-Aldrich, catalog number: 14263 )
- Acetonitrile (LC-MS Ultra CHROMASOLV) (Sigma-Aldrich, catalog number: 14261 )
- Hexane (Sigma-Aldrich, catalog number: 296090 )
- HCl (Sigma-Aldrich, catalog number: 258148 )
- Trypan blue (Gibco, catalog number: 15250-061 )
- Phorbol myristate acetate (PMA) (Sigma-Aldrich, catalog number: P8139 )
- Pyridine (Sigma-Aldrich, catalog number: 270970 )
- Sulfur trioxide pyridine (Sigma-Aldrich, catalog number: S7556 )
- Barium acetate (Sigma-Aldrich, catalog number: 243671 )
- D-Glucose (13C6 99%) (Euriso-Top, catalog number: CLM-481 )
- RPMI culture media 1640 (Gibco, catalog number: 31800-014 )
- Glucose free RPMI media-1640 (Sigma-Aldrich, catalog number: R1383 )
- Dialyzed FBS (Hyclone, catalog number: SH30071.03 )
- Cholate buffer (see Recipes)
- Quenching mix (see Recipes)
Equipment
- Bright-line hemacytometer (Sigma-Aldrich, catalog number: Z359629 )
- Cell scraper
- Culture dish
6 well plate (Nunc, catalog number: 140685 )
Flask (T-175, catalog number: 178983 ) - Liquid nitrogen cylinder
- Nitrogen gas cylinder
- Water bath
- Refrigerators (-20, -80, 4 °C)
- Microcentrifuge (Eppendrof, model: 5418R )
- Mono spin C18 plugs (GL Sciences, catalog number: 5010-21700 )
- HPLC (Agilent, model: 1260 infinity ; Pump: Binary Pump VL, model: G1312C )
- HPLC column
Amino column (Polaris 5 NH2 150 * 2.0 mm)
C18 column [ZORBAX Eclipse Plus C18 (Narrow Bore 2.1 * 150 mm 5 μ)]
Cyano column (Phenomenax Luna 150 * 2.0 mm 3 μ) - Mass spectrometer (ABSciex, hybrid 4000 QTrap)
- Amber glass vials (Supelco, catalog number: 29117 )
- Glass vials (Borosil, catalog number: 9910 )
- Glass pasture pipette
- Sonicator (Branson, model: 1210 )
Procedure
- Metabolite labeling and extraction (Mehrotra et al., 2014)
- Culture cells in desired media. For the current protocol, THP1 cells were cultured in RPMI media, supplemented with 10% FCS at 37 °C, 5% CO2. Confluent cells were counted (using trypan blue) and seeded in phorbol myristate acetate (PMA) containing complete media for differentiation at a density of 0.8 million cell/media complete media. The final concentration of PMA used was 30 ng/ml complete media and differentiation was allowed for 48 h. If one wishes to perform kinetic labeling, seed multiple sets of cells, depending on time points to be covered. e.g. seed separate sets of cells to extract metabolites after -0, 1, 2, 5, 15, 30 and 60 min of label introduction. (The PMA differentiation step is for THP1 cells. Seeding for other adherent mammalian cells can be performed as is done routinely. The number of cells to be seeded for metabolite extraction to get good signals on the mass-spectroscope has to be optimized for each cell type. The signals obtained in our case were derived from 5 million cells, seeded in 3 wells of 6 well plate in a total of 6 ml PMA containing complete media.)
For labeling of cells with 13C6 glucose (or any other label), replace the media in culture dishes with fresh RPMI media supplemented with 10% dialyzed fetal calf serum one hour before introduction of label (it is important to use dialyzed serum as un-dialyzed forms are rich in small molecules including glucose). At the time of labeling, remove media from the culture dish and perform a quick wash of cells with glucose free media (the entire step should not exceed 30 sec- using 1-2 ml of media). This step is crucial to remove unlabeled glucose media from the culture dish. - Add 13C6-glucose containing RPMI media, supplemented with 10% dialyzed fetal calf serum to the cells for the required amounts of time, which in our case ranged from 0 min to 60 min as mentioned earlier. For upto 60 min of incubation, agitation is not required, however for longer incubation time, gentle agitation in breaks may become important. (Glucose concentration-2 mg labeled glucose added per ml of glucose free RPM media, supplemented with 10% FCS.) Make sure that the media temperature is 37 °C since temperature fluctuations can induce changes in the metabolic profile.
- At the completion of incubation time, immediately remove the labeled media completely and add the quenching mix- chilled (-70 °C), methanol-water (80: 20) to the cells. For our study we added 700 µl of the quenching mixture which was sufficient to cover the surface of seeded cells. There is no need to wash cells with Glucose free media before adding the quenching mix as this will only increase the exposure time of cells to labeled media (further, since we did not measure glucose on the mass spectrometer, the left over quantity of label carrying 13C glucose did not make any difference to our observations). The quenching mix may be spiked with an external standard to gauge for any losses during extraction. (Example of an of external standard is Fumaric acid-13C4, d4. which shows a peak shift corresponding to M+8 from Fumaric acid. The standard can be added to the quenching mix (80:20 methanol: water) before using it for metabolic quenching. The concentration of the standard needs to be optimized and may be used in a range from 0.1 to 100 ng/ul of quenching mix).
- Immediately place the culture dishes in -75 °C for 10 min to allow for complete metabolic quenching. This is followed by incubation on ice for 10-15 min to allow for freeze-thaw lysis of cells.
- The cells are then scraped off the culture dish on dry ice.
- Vortex for 10 min with 30 sec of votexing followed by 1 min incubaton on ice.
- Spin the lysate at 6,000 x g for 5 min at 4 °C.
- Collect supernatant and add a 200 µl of quenching mix to the pellet and vortex hard. Re-spin the tube and collect the supernatant. Repeat step.
- Pool the three supernatants obtained (steps A9-10).
- Dry the supernatants under a stream of N2 gas. 1,000 µl of extract will dry up in approximately 20 min.
- Re-suspend the dried extract in MS grade water (re-suspension volume is dependent on cell number and MS-sensitivity range. For 5 million THP1 cells we used 180 µl volume).
- Pass the samples once through the mono spin C18 columns to remove any particulate debris (this allows for greater longevity of the LC column).
- Proceed for LC-MS analysis and the samples may be further diluted with acetonitrile or other organic solvents depending on the LC column requirements. It is best to analyze the isolated metabolites on the mass spectroscope within 24 h of isolation. Extracted metabolites should be stored at -80 °C. The extracts, while processing, re-suspension or mass analysis should be maintained at 4 °C.
- Culture cells in desired media. For the current protocol, THP1 cells were cultured in RPMI media, supplemented with 10% FCS at 37 °C, 5% CO2. Confluent cells were counted (using trypan blue) and seeded in phorbol myristate acetate (PMA) containing complete media for differentiation at a density of 0.8 million cell/media complete media. The final concentration of PMA used was 30 ng/ml complete media and differentiation was allowed for 48 h. If one wishes to perform kinetic labeling, seed multiple sets of cells, depending on time points to be covered. e.g. seed separate sets of cells to extract metabolites after -0, 1, 2, 5, 15, 30 and 60 min of label introduction. (The PMA differentiation step is for THP1 cells. Seeding for other adherent mammalian cells can be performed as is done routinely. The number of cells to be seeded for metabolite extraction to get good signals on the mass-spectroscope has to be optimized for each cell type. The signals obtained in our case were derived from 5 million cells, seeded in 3 wells of 6 well plate in a total of 6 ml PMA containing complete media.)
- Lipid (fatty acids and cholesterol) labeling and extraction
- Fatty acid and cholesterol labeling
- Seed and label the cells as described in step A14. For lipid labeling experiments in our study, cells were incubated in the label carrying media for 4 h, unlike the labeling of metabolites, which was carried out for short intervals with maximum being 60 min. The cell number taken for lipid labeling experiments was 30 million, seeded in a T-175 flask.
- Completely remove the labeling media from the flask at the end of labeling time.
- Add cholate buffer to the flasks and incubate at RT for 5 min with gentle intermittent tapping. For 30 x 106 THP1 cells seeded in T-175 flasks, 3 ml of buffer was used. To collect cells efficiently, use a scraper.
- Collect the lysate and perform a hard vortex (vortex for 5 min, 30 sec vortex followed by 30 sec incubation on ice) followed by centrifugation at 3,000 x g for 10 min at 4 °C.
- Seed and label the cells as described in step A14. For lipid labeling experiments in our study, cells were incubated in the label carrying media for 4 h, unlike the labeling of metabolites, which was carried out for short intervals with maximum being 60 min. The cell number taken for lipid labeling experiments was 30 million, seeded in a T-175 flask.
- Lipid extraction: Free fatty acid isolation
Free fatty acids were obtained by following the protocol by Aveldano and Horrocks (1983) with slight modifications (Aveldano and Horrocks, 1983). For lipid extraction and handling, strictly glassware should be used. It is important to note here that nowhere during lipid extraction and handling should plastic ware be used.- Extract lipids from the cells using the Bligh and Dyer protocol in Borosil glass tubes (Bligh and Dyer, 1959). Briefly, to the each 1 ml of lysate collected in step B1d; add 3.75 ml 1:2 CHCl3: MeOH and vortex well for a minute. Then add 1.25 ml of CHCl3 and vortex well. This is followed by the final addition of 1.25 ml of water and vortexing. The tubes are then centrifuged at 1,000 rpm for 5 min at room temperature to yield a two-phase system. Carefully extract the bottom layer as the lipid containing zone using a glass pasture pipette.
- Dry extracted lipids under a stream of nitrogen gas and store in -20 °C in N2 gas atmosphere until further use. Since lipids are extremely prone to peroxidation, store dried lipids in vials topped with nitrogen gas, in dark.
- Re-suspend the dried lipids in 100 μl of MS grade water and add 1 ml of 4:1 acetonitrile: 37% (v/v) hydrochloric acid.
- Cap the vials and incubate at 90 °C for 2 h to allow for acid hydrolysis of all triacyl glycerides, allowing for the release of fatty acids.
- Cool down the extracts to room temperature and add 1 ml of hexane, followed by vortexing for 20 sec.
- Leave the samples at RT for 5 min, undisturbed, followed by centrifugation at 3,000 x g for 5 min.
- Collect the supernatant, which is the layer with hydrolyzed lipids. Perform the same procedure of hexane addition to the pellet and pool the two supernatants.
- Measure the volume of collected sample accurately. Divide it into two equal halves and dry both sets under nitrogen stream.
- After drying the first set, add 200 µl of 50: 40: 5 chloroform: methanol: water and 0.01% aqueous ammonia, vortex well and use directly for Lipid analysis.
- Use the second set for cholesterol derivatization.
- Extract lipids from the cells using the Bligh and Dyer protocol in Borosil glass tubes (Bligh and Dyer, 1959). Briefly, to the each 1 ml of lysate collected in step B1d; add 3.75 ml 1:2 CHCl3: MeOH and vortex well for a minute. Then add 1.25 ml of CHCl3 and vortex well. This is followed by the final addition of 1.25 ml of water and vortexing. The tubes are then centrifuged at 1,000 rpm for 5 min at room temperature to yield a two-phase system. Carefully extract the bottom layer as the lipid containing zone using a glass pasture pipette.
- Lipid extraction: Cholesterol derivatization
Cholesterol molecule does not ionize well in the mass spectrometer. To aid the ionization process, cholesterol is converted to cholesterol sulphate by following the protocol by Sandhoff et al. (1999).- In a fresh dry, glass amber vial, add 2.5 mg of sulfur trioxide pyridine, followed by addition of 2.5 mg of dry pyridine.
- Sonicate the contents for 10 sec in a bath sonicator using ultrasonics at room temperature..
- Add 20 µl of the solution from B3b to the vial containing the dried sample (i.e. to the sample stored at step B2j). The sample vial must be at RT. Sonicate in water bath for 10 sec.
- Leave the samples at RT for 15 min.
- At completion of incubation, add 2.1 μl of 314 mM solution of barium acetate. Sonicate for 10 sec in water bath.
- Incubate at RT for 10 min, followed by incubation at 4 °C for 60 min.
- At the completion of incubation, allow vials to come to RT. Add 120 µl of methanol and centrifuge the mix at 13,000 x g for 10 min.
- The supernatant can be directly used for mass spectrometric measurement of cholesterol.
- In a fresh dry, glass amber vial, add 2.5 mg of sulfur trioxide pyridine, followed by addition of 2.5 mg of dry pyridine.
- Fatty acid and cholesterol labeling
- Results and interpretation
As the labeled glucose moieties get metabolized in the cells, the quantity of down-stream metabolites carrying the label (formed by glucose catabolism) increases. In parallel, a decrease in their unlabeled isotopic form is observed. The phenomenon can be captured on the mass spectroscope.
There are essentially two ways of data evaluation:- Absolute metabolite quantitation and determination of formation/degradation rates
- Standard curves: Generate calibration curves on the mass spectrometer, for the metabolites to be monitored, by injecting standard solutions covering a range of concentrations.
- Determination of labeling transitions of metabolites: Analyze the mass spectrometry data obtained from the test samples, to carefully identify the labeling patterns in each of the metabolites. For example, when 13C6 Glucose is fed to THP1 cells, there are numerous possible labeling patterns (transitions) that can be obtained on the mass-spectrometer for Dihydroxy acetone phosphate (DHAP). However, if labeled glucose, catabolize via glycolysis, is the only major contributor to the molecule, then the most abundant transitions detected on the mass spectrometer will be either the completely unlabeled form or the one carrying all the labeled carbons.

Figure 1. The two most abundant labeling patterns (transitions) for DHAP obtained on exposing cells to 13C6 Glucose for short durations. T1: completely unlabeled form of the molecule (12C), T2: all labeled carbon atoms (13C) - Determination of concentration of each of the transitions: Use the calibration curve to determine the concentration of each of the transition for all the metabolites under study in the test samples. The procedure should be done for all time points post-labeling.
- It is important to know the volume of sample prepared (re-suspension volume for the extract from 5 million cells, after drying under N2 gas) and the volume of sample injected into the Mass-spectrometer in order to determine the concentration of the metabolites. The net amount of metabolite in the sample, can be obtained by adding the concentrations of both 13C and 12C transitions
- Calculate rate of labeling: For each of the metabolites, determine the rate of label incorporation (or unlabeled pool’s depletion) by calculating the slope of the linear part of the concentration graph , following the below mentioned steps:
- By means of Z score by MAD (used for gauging the stability point), determine the stable 13C and 12C concentrations achieved by the isotopic forms after introduction of the label respectively. Formula used for determining the stability point:
Z-score=(xi - xm)/ MAD; MAD = median{abs(xi -xm)}
Where xi is the concentration achieved at each time point post labeling by each of the isotopes, and Xm is the median concentration achieved by the metabolite during the labeling time of 60 min.
This stability point, for 13C isotope is also the saturation concentration achieved by the metabolite. - Once the stability point (stable concentration achieved) is determined, calculate the slope of the curve followed by the metabolite in achieving the stable concentration. This gives the net rate of formation (or degradation).
The process of rate calculation has been exemplified in the section on representative data.
- Standard curves: Generate calibration curves on the mass spectrometer, for the metabolites to be monitored, by injecting standard solutions covering a range of concentrations.
- Determination of percentage of labeled isotopic form for each metabolite
Calculate the percent label incorporation into each metabolite as follows:
{(peak area of all isotopic transitions containing 13C labels)/(peak area of all isotopic transitions ie carrying 13C labels+ naturally occurring 13C isotope)}*100
- Absolute metabolite quantitation and determination of formation/degradation rates
Representative data
The nature of data obtained from kinetic flux profiling experiment, when both, the 13C and 12C isotope, are traced over labeling time, typically looks like the data set in table 1 (plotted in Figure 2).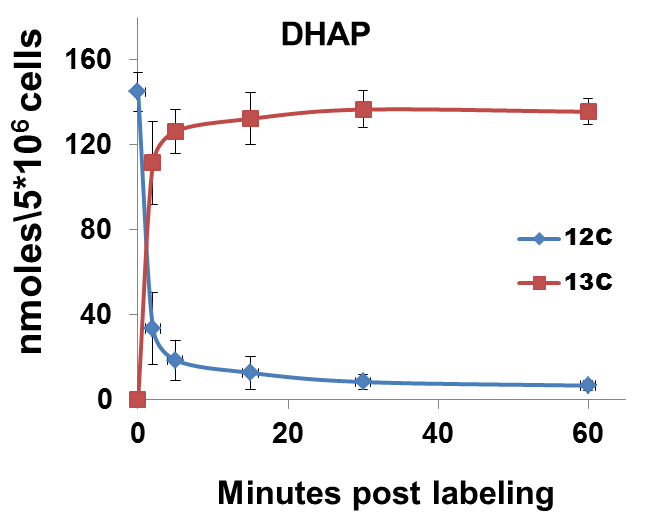
Figure 2. 13C label incorporation and corresponding reduction in 12C isotope of DHAP upon introduction of labeled media
Table 1. Table representing the values plotted in Figure 1 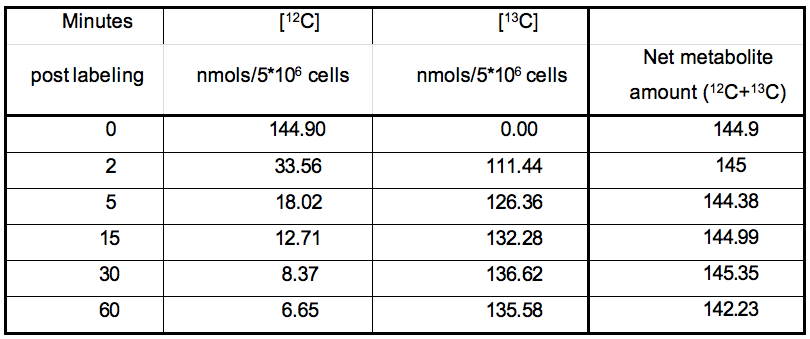
It can be noted from the data set that in metabolic pathways with high activity, a depletion of the 12C isotope occurs, with a concomitant increase in the 13C isotope quantity. The net amount of the metabolite (a sum of the 13C and 12C concentrations) remains relatively constant over experiments of short durations such as those represented here. This can be used as a thorough check of the data quality since, little or no changes should be expected in total amount of a metabolite under constant conditions during experiments with short labeling times.
Certain metabolic pathways, which are not very active, may not show active depletion of the naturally occurring 12C isotopic pool of their metabolites, in spite of certain amount of 13C labeling. Example1: Calculation of the rate of 13C label incorporation for DHAP (concentration values to be used from Table 1):
Step1: Median (Xm) = 132.28; obtained by calculating the median of 111.44,126.36, 132.28, 136.62, 135.58)
Step 2: Xi-Xm
Table 2. Calculation of Xi-Xm using values from Table 1 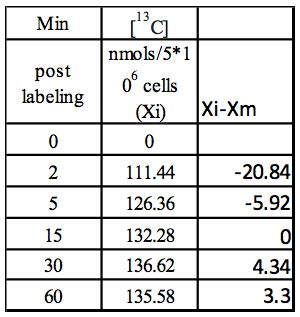
Step 3: Abs(Xi-Xm)
Table 3. Calculation of the absolute values of Xi-Xm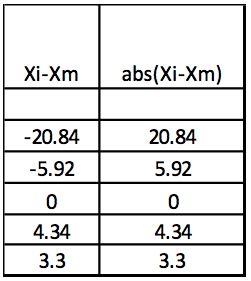
Step 4: MAD: Median of Abs(Xi-Xm) = 5.13
Step 5: Z-score
Table 4. Calculation of the Z-score for each concentration value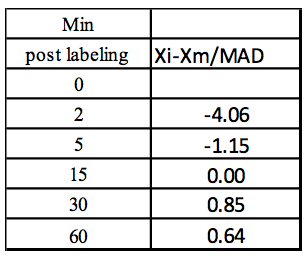
Notes
- During label switch and washes, the media should be constantly maintained at 37 °C. Fluctuations in media temperature cause variations in metabolite profiles.
- While seeding cells and labeling, make sure that the incubator surface is uniform and the media is not accumulating in one zone in the culture dish.
- During long labeling times (exceeding 1 h), it is advisable to gently swirl the culture dishes every 30 min to aid in uniform distribution of media.
- In order to account for cell loss, it is best to seed cells in a parallel plate and perform similar washes as in the lot to be extracted. Instead of adding methanol-water to this set, trypsinize cells and count to get an estimate of the cell number.
Recipes
- Cholate buffer
0.1 M potassium phosphate
0.05 NaCl
5 mM cholic acid
0.1% triton - Quenching mix
80:20 methanol: water
Acknowledgments
This work was performed as part of the SysTB consortium that is supported by a grant from Department of Biotechnology (DBT)- Government of India. PM is a Senior Research Fellowship recipient from Council of Scientific and Industrial Research-Government of India.
References
- Aveldano, M. I. and Horrocks, L. A. (1983). Quantitative release of fatty acids from lipids by a simple hydrolysis procedure. J Lipid Res 24(8): 1101-1105.
- Bligh, E. G. and Dyer, W. J. (1959). A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37(8): 911-917.
- Mehrotra, P., Jamwal, S. V., Saquib, N., Sinha, N., Siddiqui, Z., Manivel, V., Chatterjee, S. and Rao, K. V. (2014). Pathogenicity of Mycobacterium tuberculosis is expressed by regulating metabolic thresholds of the host macrophage. PLoS Pathog 10(7): e1004265.
- Sandhoff, R., Brügger, B., Jeckel, D., Lehmann, W. D. and Wieland, F. T. (1999). Determination of cholesterol at the low picomole level by nano-electrospray ionization tandem mass spectrometry. J Lipid Res 40(1): 126-132.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Mehrotra, P., Saquib, N. and Rao, K. V. S. (2015). 13C Kinetic Labeling and Extraction of Metabolites from Adherent Mammalian Cells. Bio-protocol 5(8): e1447. DOI: 10.21769/BioProtoc.1447.
- Mehrotra, P., Jamwal, S. V., Saquib, N., Sinha, N., Siddiqui, Z., Manivel, V., Chatterjee, S. and Rao, K. V. (2014). Pathogenicity of Mycobacterium tuberculosis is expressed by regulating metabolic thresholds of the host macrophage. PLoS Pathog 10(7): e1004265.
Category
Biochemistry > Lipid > Lipid isolation
Cell Biology > Cell metabolism > Lipid
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.