- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

TUNEL Assay in Kiwifruit Stigmatic Arms



Published: Vol 5, Iss 7, Apr 5, 2015 DOI: 10.21769/BioProtoc.1434 Views: 11901
Reviewed by: Arsalan DaudiKanika GeraAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling (TUNEL) is a method for detecting DNA fragmentation by labelling the 3' terminal end of nucleic acids. This method can be used both in animal and plant tissues. In animal tissues, the use of Proteinase K is sufficient for permeabilizing the cells and to obtain optimal labelling, but in plant tissues, the presence of the cell wall, does not allow proper labelling. For this reason, we carried out several modifications to the original TUNEL protocol (ApoAlert® DNA Fragmentation Assay Kit, Clontech) to obtain an optimal labelling. These modifications were additional treatments with cellulase, Triton X-100 and Proteinase K. Also, we describe the optimization of the positive controls by adjusting the units of DNase used. The PI concentration for counterstaining has been also specifically adjusted to avoid excessive background noise and hence to correctly observe both labeled and unlabelled nuclei. This work also describes an additional protocol to collect, store and include samples (specifically stigmatic arms) in such a way that they do not interfere with the TUNEL labelling.
Keywords: Programmed Cell DeathMaterials and Reagents
- Acetic acid (CH3COOH) (Panreac Applichem)
- Acetone [(CH3)2CO] (Panreac Applichem)
- ApoAlert® DNA Fragmentation Assay Kit (Clontech, catalog number: 630107 )
- (3-Aminopropyl) triethoxy-silane (APTES) (C9H23NO3Si) (Sigma-Aldrich, catalog number: 440140 )
- Cellulase Onozuka RS (Duchefa Biochemie, catalog number: C8003 )
- Citifluor Solid Mountant kit (Agar Scientific, catalog number: AGR1326 )
- Clear nail polish
- Deionized H2O (Milli-Q H2O)
- Distilled water
- Disodium hydrogen phosphate (Na2HPO4) (Duchefa Biochemie)
- Ethylenediaminetetraacetic acid (EDTA) (C10H16N2O8) (Duchefa Biochemie)
- Ethanol (C2H5OH) (Panreac Applichem)
- Formaldehyde (HCHO) (36.5-38%) (Panreac Applichem, catalog number: 48 131328 )
- Monobasic potassium phosphate (KH2PO4) (Duchefa Biochemie)
- Paraffin (Panreac Applichem, catalog number: 253211 )
- Paraformaldehyde [(HCHO)n] (Panreac Applichem, catalog number: 141451 )
- Potassium chloride (KCl) (Duchefa Biochemie)
- Propidium iodide (PI) (Sigma-Aldrich, catalog number: P4864 )
- Proteinase K (Sigma-Aldrich, catalog number: P4850 )
- Recombinant DNase I (RNase-free) (TAKARA BIO, catalog number: 2270A )
- Sodium chloride (NaCl) (Duchefa Biochemie)
- Sodium hydroxide (NaOH) (Panreac Applichem)
- Sulfuric acid (H2SO4) (Sigma-Aldrich)
- Tertiary butyl alcohol/2-Methyl-2-Propanol (TBA) [(CH3)3COH] (Panreac Applichem, catalog number: 141903 )
- Tris (hydroxymethyl) aminomethane hydrochloride (Tris-HCl) [NH2C(CH2OH)3·HCl] (Duchefa Biochemie)
- Triton X-100 (Sigma-Aldrich)
- Xylene [C6H4(CH3)2] (Panreac Applichem)
- Fixing solution (see Recipes)
- TBA solutions (see Recipes)
- TBA:paraffin (see Recipes)
- Formulated water (see Recipes)
- APTES-coated slides (see Recipes)
- 1x PBS buffer (see Recipes)
- 4% formaldehyde in 1x PBS buffer (see Recipes)
- 2% Cellulase Onozuka in 1x PBS buffer (see Recipes)
- 0.5% Triton X-100 in 1x PBS buffer (see Recipes)
- Tris-HCl (see Recipes)
- EDTA (see Recipes)
- 100 mM Tris-HCl (see Recipes)
- Proteinase K solution (see Recipes)
- 1x DNase I buffer (see Recipes)
- 1,500 U/ml DNase I (see Recipes)
- PI solution (see Recipes)
- Anti-Fade reagent (see Recipes)
Equipment
- 15 ml tubes
- 100 ml darkened bottles
- 100 ml darkened glass bottles
- 100 ml glass flasks
- 200 ml glass Coplin jars
- 250 ml glass jars
- Autoclave
- Filters sterile Filtropur S 0.2 (SARSTEDT AG)
- Flat surface (wall tile)
- Forceps
- Fume hood
- Glass coverslips
- Glass-covered trays
- Laboratory oven
- Leica TCS-SP2 confocal microscope
- Leuckart's bars for paraffin block preparation (ICT, SL., catalog number: 101 20996051645 )
- Magnetic stirrer with heating plate
- Microtome (Shibuya optical)
- Microtome Blades Accu-Edge® low-profile (Sakura)
- Paint brush
- pH-meter
- Scalpel with sterile blades
- Slides
Software
- LCS Software for Leica TCS-SP2 confocal microscope
Procedure
- Flower collection
We collect flowers from mature female vines of kiwifruit, Actinidia chinensis var deliciosa (A.Chev.) A.Chev.) cv. ʻHaywardʼ growing in a commercial orchard located in north-western Spain (Galicia) and operated by Kiwi Atlántico S. A.
- Prepare plant material immediately after collection by detaching petals, sepals and stamens using a scalpel.

Figure 1. Plant material. A. Kiwifruit female flower. B. Female pistil (ovary 124 with stigmatic arms). C. A female stigmatic arm detached from pistil.
- Submerge completely five pistils (ovary and stigmatic arms) in a 100 ml glass flask containing 60 ml fixing solution for 24 h at 4 °C (William et al., 1999).
- Remove fixing solution, add 60 ml 75% ethanol. Store at 4 °C until use.
- Prepare plant material immediately after collection by detaching petals, sepals and stamens using a scalpel.
- Paraffin embedding stigmatic arms
- Put 5 stigmatic arms, each from a different flower, in a 15 ml tube.
- Add 5 ml of TBA V (recipe II) solution to the samples and incubate 3 h at room
temperature.
- Remove TBA V, add 5 ml TBA IV to the samples, and incubate overnight at
room temperature.
- Remove TBA IV, add 5 ml TBA III to the samples, and incubate for 70 min at
room temperature.
- Remove TBA III, add 5 ml TBA II to the samples, and incubate for 80 min at
room temperature.
- Remove TBA II, add 5 ml TBA I to the samples, and incubate for 100 min at
room temperature.
- Remove TBA I, add 5 ml pure TBA to the samples, and incubate for 85 min at
38 °C in a laboratory oven.
- Replace TBA with fresh TBA and incubate for 45 min at 38 °C in a laboratory
oven.
- Replace TBA with fresh TBA and incubate overnight at 38 °C in a laboratory
oven.
- Remove the TBA, add 5 ml TBA: paraffin (previously maintained at 60 °C in a
laboratory oven) and incubate for 24 h at 60 °C in a laboratory oven.
- Replace TBA: Paraffin with fresh one and incubate for 24 h at 60 °C in a laboratory oven.
- Remove the TBA: Paraffin add 5 ml paraffin (previously maintained at 60 °C) to the samples and incubate for 7 h at 60 °C in a laboratory oven.
- Replace the paraffin with fresh paraffin and incubate for 5 days at 60 °C in a laboratory oven.
- Put a thin layer of liquid paraffin into Leuckart bar. Place the samples arranged into horizontal position before the paraffin solidifies and completely cover with additional liquid paraffin. Two samples per mold can be prepared. Allow solidify at least 3-4 h.
- Label the paraffin blocks indicating the stigmatic arm orientation, and store them at room temperature until use.
- Put 5 stigmatic arms, each from a different flower, in a 15 ml tube.
- Section preparation
- Trim the paraffin blocks with the help of a scalpel to obtain an optimal cutting surface including the sample with a small paraffin frame.
- Fix the small paraffin frame on the microtome chuck.
- Set the microtome blade and adjust the positions of the blade and the sample.
- Cut the paraffin block to obtain ribbons of 10 μm-thick sections containing serially sectioned stigmatic arms. Pick up the sections with forceps or paint brush and store them in a glass-covered tray.
- Put a few drops of formulated water on the surface of previously prepared APTES-coated slides.
- Pick up the sections containing stigmatic arm samples from the trays with the aid of a forceps and put them onto the surface of formulated water on the APTES-coated slides (about 15-18 sections per slide).
- Leave the sections overnight at 30 °C in a laboratory oven to allow the thin sections be expanded and fully attached to the slides.
- Store slides at room temperature.
- Trim the paraffin blocks with the help of a scalpel to obtain an optimal cutting surface including the sample with a small paraffin frame.
- Sample preparation for microscopic detection
This procedure is carried out using the ApoAlert® DNA Fragmentation Assay Kit. We usually handle four slides at the same time in each protocol session. From them, two slides contain experimental samples, and the other two are controls, one negative and the other positive. For this later one (positive control), the use of dedicated laboratory material (glass coplin jar, wall tile, forceps, etc.) is mandatory.
Steps from two to eleven, and from twenty-six to twenty-nine follow the ApoAlert® DNA Fragmentation Assay Kit protocol (steps 1-10, and 12-14, respectively).
- Incubate the slides at 60 °C in a laboratory oven for 7 min to ensure the complete removal of paraffin.
- Remove paraffin by immersing the slides in Coplin jars containing fresh xylene. Incubate at room temperature for 5 min.
- Repeat once the second step by transferring the slides to a new Coplin jar containing fresh xylene.
- Immerse the slides in Coplin jars containing fresh 100% ethanol and incubate at room temperature for 5 min.
- Rehydrate the slides by sequentially incubating them for 3 min steps at room temperature in different Coplin jars containing: 100% ethanol 95% ethanol 85% ethanol 70% ethanol 50% ethanol.
- Immerse the slides in Coplin jars containing fresh 0.85% NaCl (p/v) and incubate them at room temperature for 5 min.
- Immerse the slides in Coplin jars containing fresh 1x PBS buffer and incubate them at room temperature for 5 min.
- Fix the slides by immersing them in Coplin jars containing fresh 4% formaldehyde and incubate them at room temperature for 15 min.
Note: Let cellulase to thaw on ice to be used in step 12.
- Wash the slides by immersing them in Coplin jars containing 1x PBS buffer, and incubate them at room temperature for 5 min.
- Transfer the slides to another Coplin jar containing 1x PBS buffer, and incubate them again at room temperature for 5 min.
- Allow the liquid to drain thoroughly and place the slides on a flat surface (wall tile). The cellulase, Triton X-100 and Proteinase K treatments must be carried out with care due to risk of losing sections, which can slip away from the slides.
- Cover every section with 2% cellulase Onozuka RS. Solution must completely cover the sections. Usually this can be done with 1 ml of solution per slide.
- Cover the slides with inverted Coplin jars containing wet paper towels in the bottom to ensure high humidity (Figure 2).

Figure 2. Inverted Coplin jars containing wet paper towels in the bottom to ensure high humidity during laboratory oven incubation of slides
- Incubate at 37 °C for 90 min in a laboratory oven.
Note: Prepare 0.5 % Triton X-100 in 1x PBS buffer to be used in step 18.
- Immerse the cellulase-treated slides in Coplin jars containing 1x PBS buffer and incubate them at room temperature for 5 min.
- Repeat twice the step 15 using a new Coplin jar containing fresh 1x PBS buffer.
- Allow the liquid to drain thoroughly and place the slides on a wall tile.
- Cover every section with 0.5% Triton X-100 in 1x PBS buffer. Solution must completely cover the sections. Usually this can be done with 1 ml of solution per slide.
- Incubate for 20 min at room temperature.
Note: Prepare the 20 μg/ml Proteinase K solution to be used in step 23.
- Immerse the Triton X-100-treated slides in Coplin jars containing 1x PBS buffer and incubate them at room temperature for 5 min.
- Repeat twice the step 20 using a new Coplin jar containing fresh 1x PBS buffer.
- Allow the liquid to drain thoroughly and place the slides on a wall tile.
- Cover every section with the 20 μg/ml Proteinase K. Solution must completely cover the sections. Usually this can be done with 1 ml of solution per slide.
- Cover the slides with inverted Coplin jars containing wet paper towels in the bottom to ensure high humidity (Figure 2).
- Incubate at 37 °C for 30 min in a laboratory oven.
- Immerse the Triton X-100-treated slides in Coplin jars containing 1x PBS buffer and incubate them at room temperature for 5 min.
- Immerse the slides in Coplin jars containing 4% formaldehyde and incubate them at room temperature for 5 min.
- Repeat step 26 using a new Coplin jar containing fresh 1x PBS buffer.
- Leave the slides in fresh 1x PBS buffer, except that used to prepare the positive control.
- Prepare the positive control (with DNase) taking the slide containing the chosen sections for it and kept it separately from the other slides (experimental samples and negative control) after this point. It is necessary to use dedicated laboratory material (Coplin jars, forceps, wall tile, etc).
- Allow the liquid to drain thoroughly and place the slide on a wall tile.
- Add 1x DNase I buffer. Solution must completely cover the sections. Usually this can be done with 100 μl of solution.
- Incubate the slides at room temperature for 5 min.
- Gently tap the slides to remove liquid.
- Add 75 μl per slide of DNase I buffer containing 1,500 U/ml DNase I, as
suggested in the In situ Cell Death Detection Kit, Fluorescein. Ensure that
buffer completely covers the sections.
- Cover the slides with inverted Coplin jars containing wet paper towels in the
bottom to ensure high humidity (Figure 2).
- Incubate at 37 °C for 20 min in a laboratory oven.
- Gently tap the slide to remove the liquid.
- Immerse the slide in Coplin jars containing 1x PBS buffer for 5 min twice.
- Allow the liquid to drain thoroughly and place the slide on a wall tile.
- Incubate the slides at 60 °C in a laboratory oven for 7 min to ensure the complete removal of paraffin.
- Staining for TUNEL detection by microscopy (using the ApoAlert® DNA Fragmentation Assay Kit)
This procedure is carried out using the ApoAlert® DNA Fragmentation Assay Kit using all samples (experimental, negative and positive samples). Remember to keep the positive control separately from the other slides. Steps from one to twelve follow the ApoAlert® DNA Fragmentation Assay Kit protocol (steps 1-5, 7-9, 11-16).
- Remove the slides from the 1x PBS buffer and tap gently to remove excess liquid; place the slides on a wall tile.
- Cover the slides with 100 μl of equilibration buffer using a micropipette.
- Using forceps, gently place a piece of plastic coverslip on top of the slides to evenly spread the liquid.
- Equilibrate at room temperature for 10 min. Thaw Nucleotide Mix on ice and prepare TdT incubation buffer for all samples and controls. Protect Nucleotide Mix and TdT incubation buffer from light. Keep on ice at all times. Protect the slides from light after staining.
Use the following table (or similar) to prepare the experimental samples and positive controls:
Component
Volume
Total No. of reactions
Total volume
Equilibration buffer
45 μl
noo fslides+1
X μl
Nucleotide mix
5 μl
noo fslides+1
X μl
TdT enzyme total
1 μl
noo fslides+1
X μl
Total
51 μl
noo fslides+1
X μl
Use the following table (or similar) to prepare the negative controls:
Component
Volume
Total No. of reactions
Total volume
Equilibration buffer
45 μl
1
X μl
Nucleotide mix
5 μl
1
X μl
Milli-Q H2O
1 μl
1
X μl
Total
51 μl
1
X μl
- Using forceps, remove the plastic coverslip and gently tap the slides to remove excess liquid. Manage with care due to risk of losing sections. Carefully blot dry around the edges with tissue paper and place the slides on a wall tile.
- Gently place 50 μl of TdT incubation buffer on the slides with a micropipette.
- Using forceps, gently place a piece of plastic coverslip on top of the slides to evenly spread the liquid.
- To perform the tailing reaction, place the slides in darkness at 37 °C for 60 min in a laboratory oven. Cover the slides with inverted Coplin jars containing wet paper towels in the bottom to ensure high humidity and maintain in darkness by covering with aluminium foil.
- Using forceps, remove the plastic coverslips. Manage with care due to risk of losing sections.
- Terminate the tailing reaction by immersing the slides in Coplin jars containing 2x SSC buffer. Incubate at room temperature for 15 min.
- Wash the slides by immersing them in fresh Coplin jars containing 1x PBS buffer. Incubate at room temperature for 5 min.
- Repeat twice the step 11 using a new Coplin jar containing fresh 1x PBS buffer.
Note: Prepare 0.5 μg/ml PI to be used in step 14.
- Allow the liquid to drain thoroughly and place the slides on a wall tile.
- Cover every section with 0.5 μg/ml PI. Solution must completely cover the sections. Usually this can be done with 1 ml of solution per slide.
- Incubate at room temperature for 5 min.
- Wash the tissues by transferring the slides to fresh Coplin jars containing Milli-Q H2O and incubate at room temperature for 5 min.
- Repeat step 16 twice.
Note: Prepare the Anti-Fade reagent to be used in step 19.
- Allow the liquid to drain thoroughly and place the slides on a wall tile.
- Add 150 μl of Anti-Fade reagent per slide and cover the treated portion of the slide containing the sections with a glass coverslip.
- Seal the edges of the coverslip with clear nail polish and allow drying for at least 5-10 min. The samples can be immediately examined or stored overnight at 4 °C in darkness, for their observation just next day. We usually examine sections in a Leica TCS-SP2 confocal microscope. Fluorescence generated by incorporation of fluorescein-dUTP at the free 3ʼ-hydroxyl ends of fragmented DNA, is detected using an excitation wavelength of 488 nm for the TUNEL reaction and 561 nm for PI, with detection in the range of 492-550 nm and 581-625 nm, respectively.
- Remove the slides from the 1x PBS buffer and tap gently to remove excess liquid; place the slides on a wall tile.
Representative data
This improved protocol for TUNEL assay of DNA fragmentation in kiwifruit tissues relies on the use of an improved permeabilization of tissues by treatments with cellulase, Triton X-100, and adjusted Proteinase K, increased DNase I units (1,500 U/ml) for the generation of positive controls, and optimization of counterstaining with reduced IP concentration (0.5 μg/ml). These modifications allowed us to clearly visualize the differences between negative and positive nuclei for TUNEL (Figure 3A-C), in contrast with the results obtained using the original protocol of the ApoAlert® DNA Fragmentation Assay Kit, which was not specifically designed for plant tissues (Figure 3D-F). In the Figure 3A, TUNEL-positive nuclei can be observed as they are showing bright green fluorescence, while in the Figure 3B all present nuclei are stained in red. Finally, in the Figure 3C, all present nuclei can be observed, but those being TUNEL positive are stained in yellow whereas those TUNEL negative are stained in light red or orange. On the contrary, with the ApoAlert® kit (Figure 3D-F) since no TUNEL-positive nuclei can be observed (Figure 3D), all present nuclei can be observed in red (Figure 3E) or after counterstaining in orange (Figure 3F).
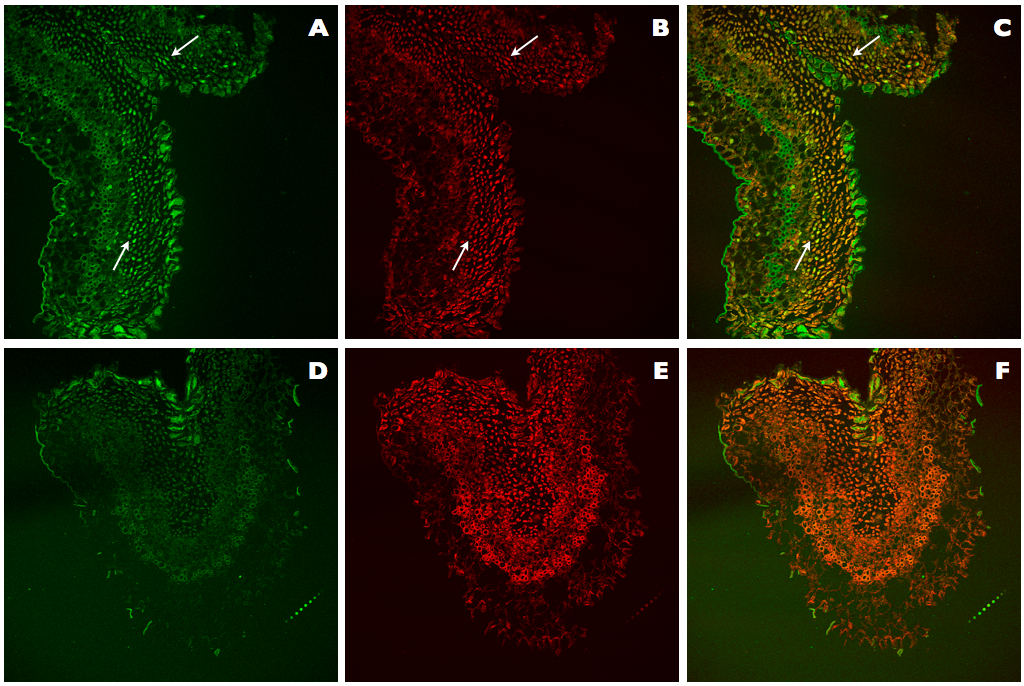
Figure 3. Comparison of TUNEL assays in positive controls of kiwifruit tissues. A-C. Positive control sections of kiwifruit stigmatic arms stained using the improved TUNEL protocol described here. D-F. Positive control sections of kiwifruit stigmatic arms stained using the ApoAlert® DNA Fragmentation Assay Kit (Clontech). A, D. Fluorescence detected at 488 nm for the observation of the TUNEL stained nuclei (arrows). B, E. Fluorescence detected at 561 nm for the observation of the IP stained nuclei (arrows). C, F. Counterstaining by simultaneous fluorescence detection at both wavelengths (488 and 561 nm) of stained nuclei with TUNEL and IP (arrows).
Recipes
- Fixing solution (100 ml)
Ethanol:acetic acid (3:1, v/v)
Mix 75 ml absolute ethanol with 25 ml acetic acid
- TBA solutions (100 ml)
TBA can solidify at room temperatureTBA I
Pure TBA
75 ml
96% ethanol
25 ml
TBA II
TBA I
73.2 ml
96% ethanol
26.8 ml
TBA III
TBA II
63.6 ml
96% ethanol
21.4 ml
H2O
15 ml
TBA IV
TBA III
57 ml
96% ethanol
21.6 ml
H2O
21.4 ml
TBA V
TBA IV
50 ml
96% ethanol
15 ml
H2O
35 ml
If this occurs, warm it in a laboratory oven at 38 °C
Stored in 100 ml darkened glass bottles at room temperature until use
- TBA:paraffin (3:1 v/v) (100 ml)
Combine 25 ml liquid paraffin with 75 ml pure TBA
The paraffin should be at 60 °C
Mix should be liquid to use
Store in 250 ml glass jar at room temperature until use
- Formulated water (100 ml)
Combine 3 ml formaldehyde 36.5-38% with 97 ml distilled water
- APTES-coated slides
Prepare 2% APTES in acetone (v/v)
Immerse the slides in 2% APTES for 3 min at room temperature
Immerse the slides in acetone for 2 min at room temperature
Repeat twice
Immerse the slides in distilled water for 1 min at room temperature
Allow to air dry (at least 24 h)
Stored at room temperature until use
- 1x PBS buffer (pH 7.4, 1 L)
10x PBS buffer (pH 7.4, 1L)
Dissolve reagents in 800 ml Milli-Q H2ONaCl
80 g
137 mM
KCl
2.2 g
3 mM
Na2HPO4
14.4 g
10 mM
KH2PO4
2.4 g
2 mM
Adjust pH
Complete volume to 1 L
Autoclave
Combine 100 ml 10x PBS buffer and 900 ml Milli-Q H2O
- 4% formaldehyde in 1x PBS buffer (pH 7.4, 1 L)
Dissolve 40 g of paraformaldehyde in 500 ml of 1x PBS buffer under a fume hood
Heat the solution at 70 °C while stirring
Add one or two drops of sulfuric acid to facilitate paraformaldehyde dilution
Complete volume to 1 L
Stored in 100 ml darkened bottles for a maximum of 1-2 weeks at 4 °C or indefinitely at -20 °C
- 2% Cellulase Onozuka in 1x PBS buffer (10 ml)
Dissolve 0.2 g of cellulase in 6 ml of 1x PBS buffer
Incubate with slow stirring overnight at 4 °C
Complete volume to 10 ml, filter sterilize (0.2 μm)
Stored at -20 °C until use
- 0.5% Triton X-100 in 1x PBS buffer (10 ml)
Dissolve 50 μl of Triton X-100 in 9.95 ml of 1x PBS buffer
Use the solution freshly prepared
- Tris-HCl (0.5 M, pH 8.0) (100 ml)
Dissolve 7.87 g of Tris-HCl in 100 ml distilled water
Adjust pH with NaOH
Autoclave and store at room temperature
- EDTA (0.2 M, pH 8.0) (100 ml)
Dissolve 5.84 g of EDTA in 100 ml distilled water
Adjust pH with NaOH
Autoclave and store at room temperature
- 100 mM Tris-HCl (pH 8.0), 50 mM EDTA (3 ml)
Combine 2 ml Tris-HCl buffer (0.5 M, pH 8.0) with 1 ml EDTA (0.2 M, pH 8.0)
- Proteinase K solution (20 μg/ml) (1 ml)
Combine 2 μl of 10 mg/ml Proteinase K with 998 μl of 100 mM Tris-HCl (pH 8.0), 50 mM EDTA
- 1x DNase I buffer (100 μl)
Dilute 10 μl10x DNase I buffer with 90 μl Milli-Q H2O
- 1,500 U/ml DNase I (75 μl)
Combine 45 μl Milli-Q H2O, 22.5 μl Recombinant DNase I and 7.5 μl 10x DNase I buffer
- PI solution (0.5 μg/ml in PBS buffer) (5 ml)
Stock: 1 mg/ml PI: Dissolve 10 mg of PI in 10 ml 1x PBS buffer
Store the stock at 4 °C in darkness
0.5 μg/ml PI solution: 2.5 μl stock and 5 ml 1x PBS buffer
- Anti-Fade reagent (1 ml)
Combine 900 μl PVA solution (Citifluor Solid Mountant kit) and 100 μl AF100 (Citifluor Solid Mountant kit)
Acknowledgments
This research was supported by the Xunta de Galicia (Regional Government of Galicia, Spain; project PGIDIT 04RAG291002PR). We thank Kiwi Atlántico S. A. for providing the plant material. We thank M. S. Costa for her expert photographic assistance, and B. Lueiro for her technical assistance. This is a contribution of the Interuniversity Research Group in Biotechnology and Reproductive Biology of Woody Plants (BioVitAc Research group, code 09IDI1705).
References
- ApoAlert® DNA Fragmentation Assay Kit User Manual (Clontech, catalog number: 630107)
- Ferradas, Y., Lopez, M., Rey, M. and Gonzalez, M. V. (2014). Programmed cell death in kiwifruit stigmatic arms and its relationship to the effective pollination period and the progamic phase. Ann Bot 114(1): 35-45.
- In Situ Cell Death Detection Kit, Fluorescein (Roche Diagnostics, catalog number: 11 684 795 910)
- Williams, J. H., Jr., Friedman, W. E. and Arnold, M. L. (1999). Developmental selection within the angiosperm style: using gamete DNA to visualize interspecific pollen competition. Proc Natl Acad Sci U S A 96(16): 9201-9206.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Ferradás, Y., López, M., Testillano, P., Rey, M. and González, M. V. (2015). TUNEL Assay in Kiwifruit Stigmatic Arms. Bio-protocol 5(7): e1434. DOI: 10.21769/BioProtoc.1434.
Category
Plant Science > Plant molecular biology > DNA > DNA modification
Molecular Biology > DNA > DNA labeling
Cell Biology > Cell imaging > Fixed-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.