- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Extraction of Small RNA and qPCR Validation of miRNAs in Vigna mungo



Published: Vol 5, Iss 5, Mar 5, 2015 DOI: 10.21769/BioProtoc.1417 Views: 11473
Reviewed by: Samik BhattacharyaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2014

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Small RNAs like microRNAs (miRNAs), small interfering RNAs (siRNAs) and other noncoding RNAs including snRNA and snoRNA have tremendous impact on eukaryotic gene regulation. Extraction of high quality small RNAs is an important prerequisite for experimental analyses of miRNAs. This will prevent RNA degradation and remove associated contaminations including polyphenols, polysaccharides and other secondary metabolites. In this protocol we describe a simple way to isolate small RNAs from the leaf tissues of Vigna mungo combining the protocols of two commercially available kits with some modifications.
Keywords: MicroRNAMaterials and Reagents
- Vigna mungo MYMIV-resistant recombinant inbred line, VMR84
- mirPremier microRNA isolation kit (Sigma-Aldrich, catalog number: SNC10 )
- Mir-X miRNA FirstStrand synthesis and SYBR qRT-PCR kit (Takara Bio Company, Clontech, catalog number: 638314 )
- 2-mercaptoethanol (Sisco Research Laboratories, catalog number: 1324196 )
- Ethanol (Merck, catalog number: K41540783 )
- Liquid nitrogen and dry ice
- RNase free water (Bangalore Genei, catalog number: 612151181001730 )
- Agarose (Sisco Research Laboratories, catalog number: 0140229 )
- Ethidium bromide (Sisco Research Laboratories, catalog number: 054817 )
- 1x TAE buffer (see Recipes)
- Ethidium bromide stock (see Recipes)
Equipment
- Thermocycler (DNA Engine Cycler, model: PTC-200 )
- Real-time qPCR (Biorad iQ5 Real-Time PCR Detection System)
- Centrifuge (Thermo Fisher Scientific, model: MicroCL21 )
- Electrophoresis apparatus (Bangalore Genei)
- Heat block or water bath
- Mortar and pestle
- NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific)
Procedure
- Plant material and growth conditions
Vigna mungo recombinant inbred line, VMR84, was used for isolation of small RNA.
- Mature seeds were surface sterilized with 0.1% HgCl2 for 10 min and rinsed twice in deionized water.
- Surface sterilized seeds were germinated in moistened filter papers at 28 ± 1 °C and 70% relative humidity and 16 h light and 8 h dark.
- Germinated seeds (after incubation of three days) were then transferred into sterile soil mix and grown in a greenhouse at 25 ± 1 °C. After 21 days of seedling growth (at 16 h light and 8 h dark), young trifoliate leaves were collected, frozen in liquid nitrogen and stored at -80 °C until RNA isolation.
- Mature seeds were surface sterilized with 0.1% HgCl2 for 10 min and rinsed twice in deionized water.
- Small RNA isolation
- Small RNA was isolated from Vigna mungo tissues using mirPremier microRNA isolation kit according to the manufacturer’s instructions with some modifications.
- The harvested leaf tissue was grounded to a fine powder in liquid nitrogen using a mortar and pestle.
- 750 μl of the lysis buffer was added to the frozen tissue powder (80 mg), vortexed for 2 min and incubated at 55 °C for 5 min.
- The sample was then centrifuged at maximum speed for 5 min to remove cellular debris, genomic DNA, and large RNA.
- The lysate supernatant (800 μl approx.) was filtered through the filtration column and the flow through was mixed with 100% ethanol and mixed immediately.
- The lysate was then passed through binding column and centrifuged at a maximum speed for 30 sec and the flow-through liquid was decanted.
- After binding, the column was first washed with 700 μl of 100% ethanol and centrifuged at 14,000 x g for 30 sec and again the flow-through was discarded.
- The second wash was done by adding 500 μl of binding solution into the column and centrifuged at maximum speed (14,000 x g) for 1 min.
- Subsequently 500 ml of the ethanol-diluted wash solution 2 was added to the column for a third wash. After centrifugation at maximum speed (14,000 x g) for 30 sec, the flow-through was discarded.
- A second wash with ethanol-diluted wash solution 2 (500 ml) was performed and the flow-through liquid was discarded.
- Next the column was dried by centrifuging at maximum speed (14,000 x g) for 1 min. The column-tube assembly was carefully removed from the centrifuge to avoid splashing of the residual flow-through liquid to the dried column.
- Small RNA was eluted from the column using 50 ml elution solution and by centrifugation at 16,000 x g and the process was repeated to improve small RNA yield. The purified RNA was stored at -20 °C.
- Quantitative and qualitative analyses of RNA were done by NanoDrop with 1 μl of the sample followed by agarose gel electrophoretic separation. The ratio of absorbance at 260 to 280 nm, calculated by (A260 - A320)/(A280-A320), is typically between 1.8 and 2.2.
- Small RNA was isolated from Vigna mungo tissues using mirPremier microRNA isolation kit according to the manufacturer’s instructions with some modifications.
- First-strand cDNA synthesis
Small RNAs were polyadenylated and reverse transcribed using the Mir-X miRNA First-Strand Synthesis kit following manufacturer’s instructions.
- Briefly, 5 μl mRQ buffer (2x), 5 μg RNA and 1.25 μl mRQ enzyme was mixed in a reaction volume of 10 μl and incubated in a thermocycler for 1 h at 37 °C, then terminate at 85 °C for 5 min to inactivate the enzymes.
- After reverse transcription, the cDNA was diluted by adding 90 μl nuclease free water to bring the total volume to 100 μl. The reverse transcribed cDNA is now ready for the miRNA quantification.
- Briefly, 5 μl mRQ buffer (2x), 5 μg RNA and 1.25 μl mRQ enzyme was mixed in a reaction volume of 10 μl and incubated in a thermocycler for 1 h at 37 °C, then terminate at 85 °C for 5 min to inactivate the enzymes.
- Quantification of miRNA by qPCR
Quantitative real time PCR was done using Mir-X miRNA qPCR SYBR Kit. The methodology is as follows:
- Designing of primers for qPCR: It is recommended to use the entire sequence of mature miRNA of desired or related plant species (available in mirBase) as miRNA specific, 5' primer. The 3' primer for qPCR is the mRQ 3' primer supplied with the kit.
- Amplification of miRNA by qPCR.
- The delta-delta Ct method was used to quantify the presence of each miRNA relative to the level of U6 snRNA, an internal control. For this experiment, qPCR amplification of U6 snRNA was done for each cDNA samples in duplicates. Additionally, inclusion of appropriate no template controls (NTC) for each primer set was performed to confirm absence of genomic DNA contamination.
- For each qPCR reaction, 20 μl PCR reaction mixture was prepared comprising of 1x SYBR advantage premix, 1x ROX dye, 0.2 mM of both forward and reverse primers and 50 ng of the first‐strand cDNA. For U6 reaction, the forward and reverse primers of U6, provided with the kit, were used.
- qPCR reactions were incubated in a 96 well plate at 95 °C for 2 min, followed by 40 cycles of 95 °C for 10 sec and 60 °C for 20 sec. Amplification cycles were followed by a melting curve analysis ranging from 56 to 95 °C, with 0.5 °C temperature increasing at every 10 sec. Melting curve for each amplicon was observed carefully to confirm the specificity of the primers used. The threshold cycle (Ct) values were recorded.
- The delta-delta Ct method was used to quantify the presence of each miRNA relative to the level of U6 snRNA, an internal control. For this experiment, qPCR amplification of U6 snRNA was done for each cDNA samples in duplicates. Additionally, inclusion of appropriate no template controls (NTC) for each primer set was performed to confirm absence of genomic DNA contamination.
- Data analysis and calculating miRNA levels using the Delta-Delta Ct (or ddCt) method.
The ddCt method provides a measure of the relative levels of a miRNA between two samples by comparing them to a normalization standard. Here the miRNA and the U6 RNA are amplified for each sample to determine the Ct value. This allows relative levels to be determined using the ddCt calculation.
The relative copy number is calculated using the ddCt method:
[miR-X] Leaf/[miR-X]Stem: 2–dCt(Leaf)/ 2–dCt(Stem)
However, precision of this method of quantification depends on the amplification efficiency of the primer pairs that should near 100% otherwise quantification will not be accurate.
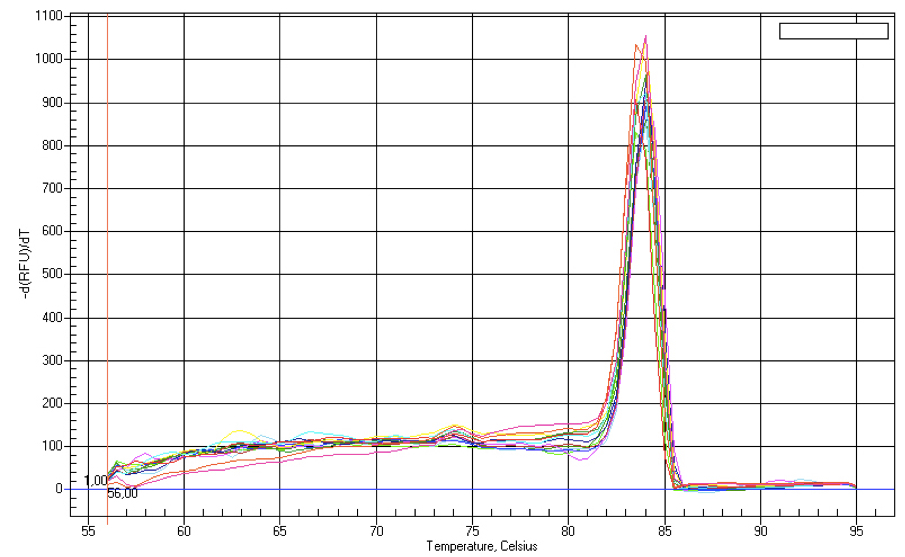
Figure 1. Melt curve analysis of mir156 produced during qPCR analysis. The graph shows a plot of negative derivative of fluorescence versus temperature (°C) for miRNA amplification. Presence of a single peak denotes nonspecific amplification of a single product.
- Designing of primers for qPCR: It is recommended to use the entire sequence of mature miRNA of desired or related plant species (available in mirBase) as miRNA specific, 5' primer. The 3' primer for qPCR is the mRQ 3' primer supplied with the kit.
Recipes
- 1x TAE (1 L)
242 g Tris base (MW 121.1), 57.1 ml Glacial acetic acid 100 ml 0.5 M EDTA was added and mixed in 600 ml of ddH2O and the final volume was adjusted to 1 L.
Next 20 ml of 50x TAE was added to 980 ml of ddH2O to prepare 1 L of 1x TAE (pH 8.3 at 25 °C).
- Ethidium bromide stock
1 g of ethidium bromide was dissolved in 100 ml of ddH2O to prepare a stock of 10 mg/ml.
Acknowledgments
The original version of this protocol was described in Paul et al. (2014). This work was supported by the Council of Scientific and Industrial Research, New‐Delhi, India for the Emeritus Scientist’s Project [Sanction No. 21 (0884)/12/EMR‐II].
References
- Paul, S., Kundu, A. and Pal, A. (2014). Identification and expression profiling of Vigna mungo microRNAs from leaf small RNA transcriptome by deep sequencing. J Integr Plant Biol 56(1): 15-23.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Paul, S., Kundu, A. and Pal, A. (2015). Extraction of Small RNA and qPCR Validation of miRNAs in Vigna mungo. Bio-protocol 5(5): e1417. DOI: 10.21769/BioProtoc.1417.
Category
Plant Science > Plant molecular biology > RNA > RNA interference
Plant Science > Plant molecular biology > RNA > qRT-PCR
Molecular Biology > RNA > RNA interference
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.