- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Conceptual Outline for Omics Experiments Using Bioinformatics Analogies

Published: Vol 5, Iss 3, Feb 5, 2015 DOI: 10.21769/BioProtoc.1387 Views: 13435
Reviewed by: Arsalan DaudiModesto Redrejo-RodriguezAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Hypothetical proteins (HP) are those that are not characterized in the laboratory and so remain “orphaned” in genomic databases. In recent times there has been a lot of progress in characterizing HPs in the laboratory. Various methods, such as sequence capture and Next Generation Sequencing (NGS), have been used to rapidly identify HP functions and their encoded genes. Applications and methods, such as the isolation of single genes, are greatly facilitated by pull-down assays to characterize proteins. Furthermore, there are methods to extract proteins from either the whole cell or a subcellular fraction. But the weakness is that some assays are fairly expensive and laborious, and characterizing HP function is always imperfect. In the recent past, statistical interpretations of the in silico selection strategies have improved the identification of the most promising candidates, including those from various annotation methods, such as protein interaction networks (PIN). Given the improvements in technology that have permitted a substantial increase in computational annotation, we ask if the prediction of HP function in silico (validation of models through algorithms and data subsets) could likewise be improved. In this work, we apply a bioinformatics analogy to each step of a wet lab experiment performed to predict aspects confirming protein function. Although it may be a less bona fide approach, assigning a putative function from conservation observed in homologous protein sequences might be worthwhile to consider prior to a wet lab experiment.
Keywords: Hypothetical proteinsProcedure
Experiment steps and bioinformatics analogies
- Immunoblotting by a Coomassie stained gel and patterns of selection for the total protein or cytoplasmic, nuclear, membrane or cytoskeletal fractions.
Analogy: A log-transformation of the data using an error model to get normally distributed noise and statistical procedures can be made using MATLAB and R (Kreutz et al., 2007). The model is applicable for simulation studies and parameter estimation in systems biology for predicting functional candidates. A bioinformatics tool, named aLFQ supports this kind of analogy where one can estimate proteomic data obtained from MS/MS, further enabling error estimation using automatic data (Rosenberger et al., 2014). Availability: Through R/CRAN (http://www.cran.r-project.org). The raw data for such analyses can be obtained from UniProt or Protein Atlas. - Genomic shotgun DNA fragments hybridized to the exome library; PCR amplification and Streptavidin beads. For example, the PCR amplification step involves finding several polymorphisms along the genome (SNP genotyping, etc.). In particular, biotinylated primer is essential for each SNP for capture of single-stranded DNA (ssDNA) template for the assay to be complete. Although alternative strategies have steadfastly been developed for pyrosequencing, the method covers all phases from PCR amplification to ssDNA template capture within pyrosequencing (Royo et al., 2007).
Analogy: Next Generation Sequencing (NGS) based annotation using HiSeq or MiSeq Illumina systems and associated materials could be used for thorough predictions (Liu et al., 2012). Galaxy frameworks can be used as an extension with machine learning based tools for sequence and tiling array data analysis. Software: HiSeq or MiSeq Illumina systems. A case study for Hi-Seq/Mi-Seq based high throughput analysis of NGS data using the Galaxy system. Please follow the help pages (see Galaxy reference). - Immunoproteomics: Kinetic analysis of antibody-peptide binding by surface plasmon resonance (SPR) is essentially used for finding antibodies against hypothetical protein candidates with high affinity. Further, a method called MALDI immunoscreening (MiSCREEN) is being used these days to screen high affinity anti-peptide antibodies (Razavi et al., 2011).
Analogy: Genetic algorithms (GAs) and swarm intelligence (SI) methods could serve as perfect replicas for feature selection methods using high-dimensional searches. Further, ant colony optimization (ACO) is used to integrate features selected on the basis of significance and applications criteria (Ressom et al., 2006). T-Coffee is a multiple sequence alignment based genetic algorithm package. These can be used to align sequences or to combine the output of favorite alignment methods into one unique alignment (Notredame et al., 2000). - Genome-wide analysis of the chromosomal distribution of co-expressed genes where one of the candidates is uncharacterized. Specificity of genes in chromosomal regions could be determined by qPCR (Boutanaev et al., 2002).
Analogy: Gene expression programming (GEP) can be used to code complex programs, which are then included in linear chromosomes of fixed length (Ferreira, 2001). These in turn could later be expressed as expression trees (ET). These ETs may further undergo mutation and recombination in predicting the function of HP candidates. One example using MATLAB demonstrates a way to find patterns in gene expression profiles, e.g. finding expressed patterns along a genome sequence (see URL: http://se.mathworks.com/help/bioinfo/ug/example-analyzing-gene-expression-profiles.html). - A major challenge in handling large scale applications for characterizing proteins using mass spectrometry, etc. is how to integrate and model the surplus of data that is produced.
Analogy: “On the fly” virtual screening where analysis is done using assembled, project-specific workflows to guide the next stages of experimentation (Pasculescu et al., 2014). The scripts can be made open-source and editable so that researchers can rapidly make enhancements in their projects. MaxQuant software package could be attributed to this analogy (Cox et al., 2009). For example, one can aim at analyzing large MS data sets and further narrow down the complex experimental designs using characterized proteins on a time series, collating them with, for instance, drug-response data. - In vivo and in vitro experimentation of cellular signaling domains
Analogy: Engineering simulations for in vivo and in vitro experimentation might be used to enable low-cost hypothesis generation and experimental design. Furthermore, in silico models can be used to develop a framework of simulations for paradigm domains, such as cancer systems biology (Bown et al., 2012). An in silico docking experiment can be perused to identify the binding residues of proteins in the open and closed conformation. Furthermore, one can get a molecular view of the system. (Degryse et al., 2008). - Many uncharacterized or HP data ultimately remain unannotated in the sequencing/biochemical information deposited from time to time.
Analogy: Aggregate different structural and functional evidence with GO relationships based on similactors (Benso et al., 2013). Further, exploit community annotation using a “wiki of uncharacterized proteins.” Please refer to Benso et al. (2013). - Antibodies vs. Aptamers. Are aptamers cost-effective when compared to antibodies for characterizing proteins (see references Aptagen and Basepairbio)? Only few analytical techniques are known to be capable of detecting minute changes with a sensitivity matching that of antibodies. The targeting of whole proteins and selection of specific residual sequences as epitopes is needed for the functional characterization of HPs. For example, a protein such as Twinkle helicase, also known as Progressive External Opthalmoplegia (PEO) in humans, is encoded by the gene C10orf2, which is similar to the GP4 helicase structure and is an interacting partner of the DNA mismatch repair protein, MLH1. A pull-down assay would resolve the purpose.
Analogy: Applying the potential role of aptamers in elucidating the function of HPs with the possibilities provided by bioinformatics for establishing a benchmark for aptamer-protein prediction methods. With these future perspectives, the role of hypothetical proteins as target molecules for diagnostics and therapies could prove to be very useful in the development of medical technology. For example, we could develop an aptamer prediction webserver, which in turn could be used for pull-down assays or label-free detection to ascertain the function of some classes of proteins, such as HPs (Suravajhala et al., 2014). Please refer to Suravajhala et al. (2014), and see the analogy below.
Aptamer Analogy
Purpose: Detailed how-to guide for implementing the bioinformatics analogy for step 8, where the role of HPs as target molecules for diagnostics and therapies could prove to be very useful in the development of medical technology. Here we use the analogy of finding better candidates (as seen pictographically in Figure 1), which could then be applied to infer function for a class of HPs.
Overview: A pull-down assay uses a small-scale affinity tag to an antibody, similar to immunoprecipitation. In the case of proteins, whose actuality, function or even interacting peers have been theoretically known but seldom experimentally established, pull-down assays can have a significant role. But can bioinformatics play a major role in lessening the scale of experimentation? The use of gene ontology functional data specific to organelles could play a major role in inferring the functions of uncharacterized proteins. For such HPs, their interacting partners remain uncharacterized as well due to the lack of feasible screening methods. Although the methods to identify the functional contexts of activity of the interacting protein have been presented, the necessary experimental boundary to characterize them explicitly does not exist. Therefore, we envisage a better predictive approach for the use of aptamers for pull-down assays or label-free detection. Application of aptamers in this research area would have immense potential as only a few analytical techniques are known to be capable of detecting minute changes with a sensitivity matching that of antibodies. Targeting whole proteins and the selection of specific residual sequences as epitopes is needed for the functional characterization of HPs, such as Twinkle helicase, also known as Progressive External Opthalmoplegia (PEO) in humans, encoded by the gene C10orf2, which is similar to the GP4 helicase structure and an interacting partner of the DNA mismatch repair protein, MLH1. We present here a step-by-step methodology to ensure this analogy is met for a biologist with little experience in bioinformatics.
Resources: Excel worksheet for transferring the annotation or even further extending the database to SQL or CSV format, and drawing software such as MS Draw or MS Publisher [for methods and software, please refer to Suravajhala and Sundararajan (2012)].
Steps- Take the HP accession in question from GenBank. Check how bona fide the accession is by identifying its related sequences, the start sites of the protein-coding regions, and whether or not it is a pseudogene. Transfer the sequence information to an Excel worksheet by employing a six-point classification scoring schema as described earlier (Suravajhala and Sundararajan, 2012).
- Find the candidate proteins that are localized to the same organelle by virtue of the interaction peers; we will be able to set aside those HPs that form an interacting pool. From the first half of the Figure 1, we show how the HP in question has its interaction peers.
- The annotation would then be transferred to the similactors approach, which will involve filtering and enrichment of PPI networks.
- Use a concrete database of aptamers that are available (Aptagen/Basepairbio). Target specifically known unknown (KU) regions and use them as putative biomarkers.
- Simulate the above list of HPs and candidate proteins from step 3 for identifying better targets from step 4.
- Analyze the results, and make a database.
- (Optional) Develop a predictive webserver based on machine learning approaches, thereby training a network of proteins and aptamers for possible and easy identification of targets.
- Take the HP accession in question from GenBank. Check how bona fide the accession is by identifying its related sequences, the start sites of the protein-coding regions, and whether or not it is a pseudogene. Transfer the sequence information to an Excel worksheet by employing a six-point classification scoring schema as described earlier (Suravajhala and Sundararajan, 2012).
Representative data (example)
In a framework for functional prediction (Figure 1), experimentally determined characteristics of the putative interaction partners are perused to make an interactome of hypothetical proteins (hypothome (Desler et al., 2014)). In this process, we suggest a role for the predicted protein in a biological context, thus complementing an interactome with the interactions with predicted proteins, in addition to retaining information on interactions, whether predicted or experimentally verified (left panel in Figure 1). This strategy is essential for characterization of predicted proteins and their interactions with existing biological pathways.
Furthermore, the electronic annotation using methods [described in Benso et al. (2013)] containing similar, yet non-interacting proteins (similactors) (right panel in Figure 1), along with the hypothome data, can be used in training datasets. However, a simulation followed by machine learning predictions can also be applied on a wide number of proteins not specific to HPs alone, thereby drawing an inference for an analogy to functional prediction.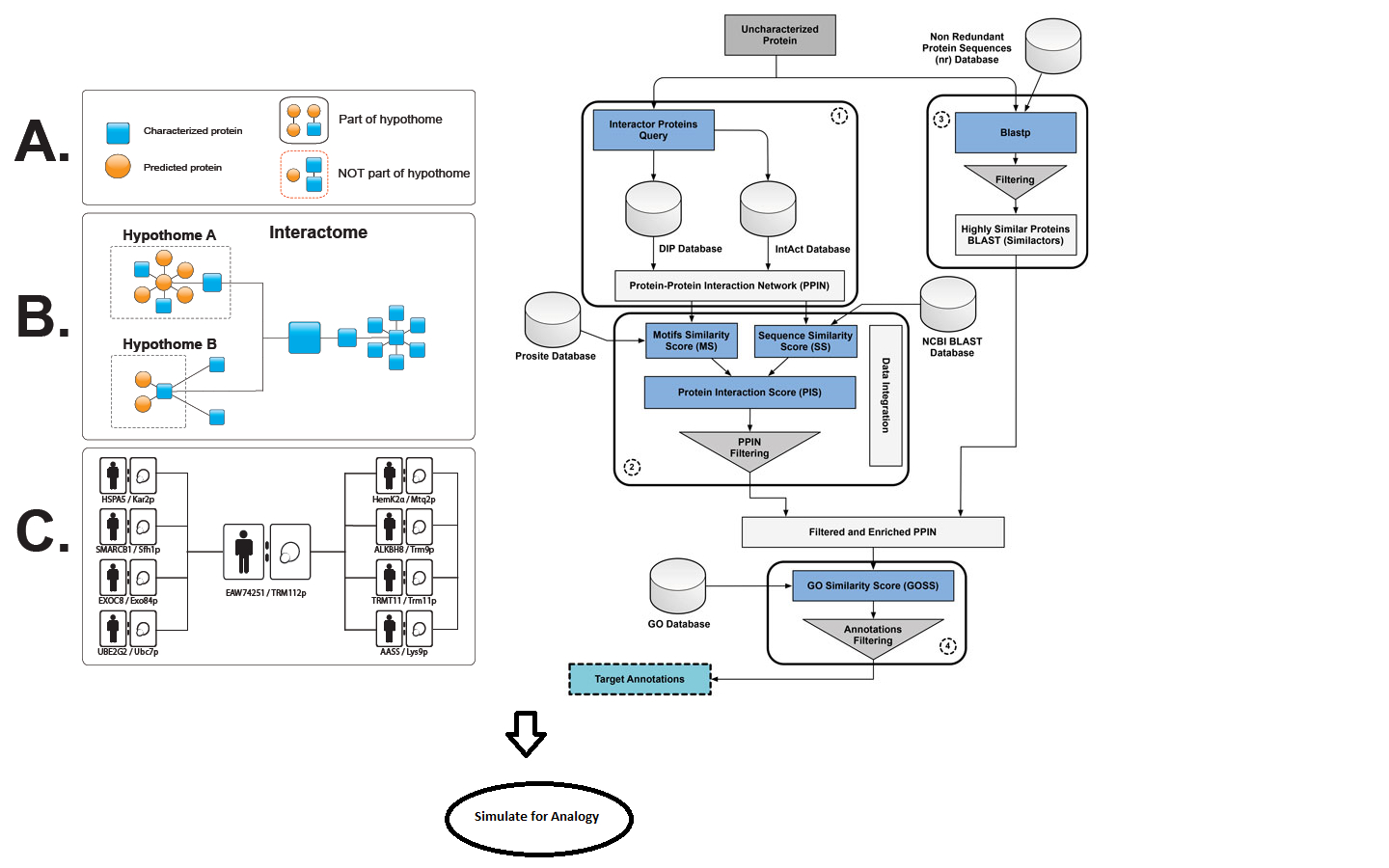
Figure 1. A framework for functional prediction. Experimentally determined characteristics of the putative interaction partners are perused to make an interactome of hypothetical proteins. Left panel: methods for making an interactome of hypothetical proteins as described by Desler et al. (2014). Right panel: electronic annotation methods described by Benso et al. (2013).
Acknowledgments
We would like to gratefully acknowledge Alfredo Benso and his colleagues for proposing similactors approach alongside hypothome. The authors received no funding whatsoever. PS would like to thank Arsalan Daudi and Fanglian He for inviting us to write this manuscript.
References
- Aptagen: http://www.aptagen.com/
- Basepairbio.com: Aptamers and Their Potential Applications at Base Pair Biotechnologies.
- Benso, A., Di Carlo, S., Ur Rehman, H., Politano, G., Savino, A. and Suravajhala, P. (2013). A combined approach for genome wide protein function annotation/prediction. Proteome Sci 11(Suppl 1): S1.
- Boutanaev, A. M., Kalmykova, A. I., Shevelyov, Y. Y. and Nurminsky, D. I. (2002). Large clusters of co-expressed genes in the Drosophila genome. Nature 420(6916): 666-669.
- Bown, J., Andrews, P. S., Deeni, Y., Goltsov, A., Idowu, M., Polack, F. A., Sampson, A. T., Shovman, M. and Stepney, S. (2012). Engineering simulations for cancer systems biology. Curr Drug Targets 13(12): 1560-1574.
- Cox, J., Matic, I., Hilger, M., Nagaraj, N., Selbach, M., Olsen, J. V. and Mann, M. (2009). A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat Protoc 4(5): 698-705.
- Desler, C., Zambach, S., Suravajhala, P. and Rasmussen, L. J. (2014). Introducing the hypothome: a way to integrate predicted proteins in interactomes. Int J Bioinform Res Appl 10(6): 647-652.
- Degryse, B., Fernandez-Recio, J., Citro, V., Blasi, F. and Cubellis, M. V. (2008). In silico docking of urokinase plasminogen activator and integrins. BMC Bioinformatics 9 Suppl 2: S8.
- Ferreira, C. (2001). Gene expression programming: a new adaptive algorithm for solving problems. Complex Systems 13(2):87-129.
- Galaxy web URL: https://galaxy.cbio.mskcc.org/.
- Ressom, H. W., Varghese, R. S. and Goldman, R. (2009). Computational methods for analysis of MALDI-TOF spectra to discover peptide serum biomarkers. In: The Protein Protocols Handbook. Springer, 1175-1183.
- Heyer, L. J., Kruglyak, S. and Yooseph, S. (1999). Exploring expression data: identification and analysis of coexpressed genes. Genome Res 9(11): 1106-1115.
- Kreutz, C., Bartolome Rodriguez, M. M., Maiwald, T., Seidl, M., Blum, H. E., Mohr, L. and Timmer, J. (2007). An error model for protein quantification. Bioinformatics 23(20): 2747-2753.
- Liu, L., Li, Y., Li, S., Hu, N., He, Y., Pong, R., Lin, D., Lu, L. and Law, M. (2012). Comparison of next-generation sequencing systems. J Biomed Biotechnol 2012: 251364.
- Notredame, C., Higgins, D. G. and Heringa, J. (2000). T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol 302(1): 205-217.
- Pasculescu, A., Schoof, E. M., Creixell, P., Zheng, Y., Olhovsky, M., Tian, R., So, J., Vanderlaan, R. D., Pawson, T., Linding, R. and Colwill, K. (2014). CoreFlow: a computational platform for integration, analysis and modeling of complex biological data. J Proteomics 100: 167-173.
- Razavi, M., Pope, M. E., Soste, M. V., Eyford, B. A., Jackson, A. M., Anderson, N. L. and Pearson, T. W. (2011). MALDI immunoscreening (MiSCREEN): a method for selection of anti-peptide monoclonal antibodies for use in immunoproteomics. J Immunol Methods 364(1-2): 50-64.
- Ressom, H. W., Varghese, R. S., Orvisky, E., Drake, S. K., Hortin, G. L., Abdel-Hamid, M., Loffredo, C. A. and Goldman, R. (2006). Ant colony optimization for biomarker identification from MALDI-TOF mass spectra. Conf Proc IEEE Eng Med Biol Soc 1: 4560-4563.
- Rosenberger, G., Ludwig, C., Rost, H. L., Aebersold, R. and Malmstrom, L. (2014). aLFQ: an R-package for estimating absolute protein quantities from label-free LC-MS/MS proteomics data. Bioinformatics 30(17): 2511-2513.
- Royo, J. L., Hidalgo, M. and Ruiz, A. (2007). Pyrosequencing protocol using a universal biotinylated primer for mutation detection and SNP genotyping. Nat Protoc 2(7): 1734-1739.
- Suravajhala, P., reddy Burri, H. V. and Heiskanen, A. (2014). Combining aptamers and in silico interaction studies to decipher the function of hypothetical proteins. Eur Chem Bull 3(8): 809-810.
- Suravajhala, P. and Sundararajan, V. S. (2012). A classification scoring schema to validate protein interactors. Bioinformation 8(1): 34-39.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Suravajhala, P. and Bizzaro, J. W. (2015). A Conceptual Outline for Omics Experiments Using Bioinformatics Analogies. Bio-protocol 5(3): e1387. DOI: 10.21769/BioProtoc.1387.
Category
Systems Biology > Interactome > Gene network
Biochemistry > Protein > Interaction > Protein-protein interaction
Biochemistry > Protein > Structure
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.