- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Purification and Sequencing of DNA Guides from Prokaryotic Argonaute



Published: Vol 4, Iss 22, Nov 20, 2014 DOI: 10.21769/BioProtoc.1293 Views: 14667
Reviewed by: Fanglian HeAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2014

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Some proteins utilize nucleic acids to guide them to complementary nucleic acid targets. One example is prokaryotic Argonaute protein, which, binds small single stranded DNA molecules as guides (Swarts et al., 2014). This protocol describes a method to purify DNA guides from these proteins. It also describes a PCR-based method to enrich the guides by PCR amplification. This methods relies on addition of a poly-A tail at the 3’-end of the ssDNA molecules by Terminal Deoxynucleotidyl Transferase (TdT), followed by ligation of a oligonucleotide to the 5’-end of the ssDNA molecule using T4 RNA ligase, and amplification by PCR. The generated dsDNA products are suitable for traditional cloning and sequencing and high-throughput sequencing. Importantly, the information which strand matches the ssDNA molecule is not lost during this process.
Materials and Reagents
- Purifying nucleic acids from proteins
- Purified protein and co-purified nucleic acids in purification buffer (e.g. TtAgo with siDNA guides)
Note: Please refer to the protocol “Expression and Purification of the Thermus thermophilus Argonaute Protein” (Swarts et al., 2014b).
- Proteinase K solution (20 mg/ml) (Life Technologies, Ambion®, catalog number: AM2548 )
- CaCl2 solution (50 μM)
- Roti phenol/chloroform/isoamyl alcohol (pH 7.5-8.0) (Carl Roth, catalog number: A156 )
- 99% ethanol
- 70% ethanol (pre-cooled to -20 °C)
- Linear acrylamide (5 mg/ml) (Life Technologies, Ambion®, catalog number: AM9520 )
Note: Alternatively, a house-made Linear Acrylamide can be used (http://www.uvm.edu/~tpdelane/lab/protocols/LinearPolyAcryl.htm).
- RNase free MQ water
- Additional materials and reagents required for analysis of purified nucleic acids (optional)
- T4 polynucleotide kinase (PNK) (New England Biolabs, catalog number M0201 )
- T4 polynucleotide kinase reaction buffer (New England Biolabs, catalog number M0201)
- T4 polynucleotide kinase (PNK) (New England Biolabs, catalog number M0201 )
- Purified protein and co-purified nucleic acids in purification buffer (e.g. TtAgo with siDNA guides)
- Enriching and preparing ssDNA molecules for sequencing
- Purified single stranded DNA nucleotides in MQ water (e.g. see Procedure part A "purifying nucleic acids from proteins")
- RNase A (DNase and protease-free) (10 mg/ml) (Thermo Fisher Scientific, catalog number: EN0531 )
- Terminal Deoxynucleotidyl transferase (TdT) (recombinant) (Life Technologies, InvitrogenTM, catalog number: 10533-073 )
- Terminal Deoxynucleotidyl transferase (TdT) buffer (Life Technologies, InvitrogenTM, catalog number: 16314-015 )
- dATP (100 mM) (Thermo Fisher Scientific, catalog number: R0141 )
- QIAquick nucleotide removal kit (QIAGEN, catalog number: 28304 )
- Primer A (5’-GAGAGAGGATCCCGAATTGTGCAGCTGTCAATCAACC-3’) (5 μM)
- T4 RNA ligase 1 (New England BioLabs, catalog number: M0204 )
- T4 RNA ligase reaction buffer (10x, supplied with T4 RNA ligase 1)
- ATP (10 mM) (supplied with T4 RNA ligase 1)
- PEG8000 (supplied with T4 RNA ligase 1)
- Primer B (5’-GAGAGAGGATCCTTTTTTTTTTTTTTTTTTTTTTTTTTVN-3’) (5 μM)
- Pfu DNA polymerase (or equivalent)
- 10x Pfu DNA polymerase buffer (or equivalent, supplied with MgCl2 or MgSO4)
- dNTPs (5 μM)
- 2% agarose gel
- Generuler low range DNA ladder (Thermo Fisher Scientific)
- GeneJET gel extraction kit (Thermo Fisher Scientific, Fermentas, catalog number: K0691 )
Note: Additional materials and reagents required only if ssDNA is not 5’-end phosphorylated: T4 polynucleotide kinase (PNK, New England Biolabs, catalog number: M0201) and T4 polynucleotide kinase reaction buffer (New England Biolabs, catalog number: M0201).
- Purified single stranded DNA nucleotides in MQ water (e.g. see Procedure part A "purifying nucleic acids from proteins")
Equipment
- Purifying nucleic acids from proteins
- Heat block
- PCR machine
- Cooled table top Eppendorf centrifuge
- Freezer (-20 °C)
- Heat block
- Enriching and preparing ssDNA molecules for sequencing
- Heat block
- PCR machine
- Table top centrifuge
- Agarose gel electrophoresis equipment
- Heat block
Procedure
- Purifying nucleic acids from proteins
- Take purified protein sample (e.g. 50-500 µl, protein concentration up to 10 μM).
- Add CaCl2 to a final concentration of 5 µM.
- Add 1/10 volume of Proteinase K solution (e.g. to 450 µl protein/CaCl2 solution, add 50 µl Proteinase K solution).
- Incubate 4 h at 65 °C in a heat block. Alternatively, small volumes (up to 200 µl) can be incubated 4 h at 65 °C in a PCR machine.
- Add Roti phenol/chloroform/isoamyl alcohol (pH 7.5-8.0) in a 1:1 ratio (e.g. to 500 µl, add 500 µl phenol/chloroform/isoamyl alcohol).
- Vortex at max speed for 5 sec.
- Centrifuge the sample for 15 min in a table top centrifuge at maximum speed.
- Carefully pipet the upper fraction (aqueous phase containing the nucleic acids) to a clean centrifuge tube. Take less than the actual upper fraction (e.g. from a 500 μl sample, take only 450 μl). This avoids mixing with the lower fraction (organic phase containing proteins) which contains the phenol which might interfere with downstream steps. Discard the lower fraction.
- If the total volume of the upper fraction is larger than 500 µl, distribute the sample equally over multiple tubes.
- Add 99% ethanol in a 2:1 ratio (ethanol:sample).
- Add 10 µl linear polyacrylamide (5 µg/µl) as carrier.
- Incubate the samples at -20 °C for at least 16 h. Small nucleic acids take longer to precipitate.
- Centrifuge in a table top centrifuge at 4 °C for 30 min at maximum speed (13,000 rpm).
- Remove all ethanol with a pipette, a small white/transparent pellet should be visible-try not to disturb the pellet.
- Add 500 µl pre-cooled -20 °C, 70% ethanol to the pellet.
- Centrifuge in a table top centrifuge at 4 °C for 15 min at maximum speed.
- Remove all ethanol with a pipette-a small white/transparent pellet should be visible-try not to disturb the pellet.
- Incubate the reaction tube at 50 °C until all ethanol is evaporated (5-10 min).
- Resuspend the nucleic acids in 50 μl RNase-free MQ water.
- Analyze nucleic acids by 32P-labeling (see Figure 1) or use for enriching and preparing ssDNA molecules for sequencing (procedure B below). For 32P-labeling, use T4 polynucleotide kinase (T4 PNK) according to the protocol provided by the manufacturer.
- Take purified protein sample (e.g. 50-500 µl, protein concentration up to 10 μM).
- Enriching and preparing ssDNA molecules for sequencing
- Add 1 µl of DNase free RNase A to 50 µl purified ssDNA.
- Incubate at RT for 10 min.
- To polyadenylate the ssDNAs, use Terminal Deoxynucleotidyl transferase according to the protocol provided by the manufacturer.
10 µl 5x TdT reaction buffer
36.5 μl ssDNA (30-100 ng, if necessary, add MQ water)
1.5 μl dATP (100 mM)
1.5 μl TdT
- Mix and incubate for 60 min at 37 °C.
- Incubate for 10 min at 70 °C to inactivate TdT.
- Purify the samples using the QIAquick nucleotide removal kit according to the manual provided by the manufacturer. Elute in 30 μl MQ water.
Notes:
- Other nucleic acid purification kits might also be suitable for purifying the nucleic acids. However, most nucleic acid purification kits do not purify nucleic acids <100 nt. Ensure the kit you use is suitable for the size of nucleic acid you are purifying.
- The next step is dependent on the presence of a phosphorylated 5’-end on your ssDNA molecules. If you are not sure if your small DNA molecule has a phosphorylated 5’-end, it is possible to phosphorylate it with T4 polynucleotide kinase according to the manual provided by the manufacturer. After 5’-end phosphorylation, purify the samples using the QIAquick nucleotide removal kit according to the manual provided by the manufacturer. Elute in 30 μl MQ water.
- Other nucleic acid purification kits might also be suitable for purifying the nucleic acids. However, most nucleic acid purification kits do not purify nucleic acids <100 nt. Ensure the kit you use is suitable for the size of nucleic acid you are purifying.
- Ligate primer A (see Notes a and e) to the phosphorylated 5’-ends of the ssDNA molecules with 3’-end poly A tails by mixing 16 μl ssDNA, 1 μl primer A, 6 μl ATP (10 mM), 6 μl 10x Reaction buffer, 30 μl 50% PEG8000 (w/v) and 1 μl T4 RNA ligase. Incubate for 16 h at 25 °C. T4 RNA ligase is used as it can ligate a 3’-end of a ssDNA strand (the primer) to a 5’-end phosphorylated ssDNA strand (the ssDNA molecules with 3’-end poly A tails).
- Purify the samples using the QIAquick nucleotide removal kit according to the manual provided by the manufacturer. Elute in 30 μl MQ water.
- To enrich the ssDNA molecules, PCR amplify the ssDNA molecule using primers A and B.
2 μl 10x Pfu polymerase buffer (containing MgCl2 or MgSO4)
14 μl ssDNA with 5’ oligonucleotide and 3’ poly A tail
1 μl primer A (5 μM)
1 μl primer B (5 μM)
1 μl dNTPs (5 μM)
1 μl Pfu polymerase
- Run a PCR
1 min at 95 °C
----------
20 cycles:
1 min at 95 °C (denaturation)
30 sec at 40 °C (annealing)
1 min at 72 °C (elongation)
----------
1 min at 72 °C
Notes:
- The annealing temperature is dependent on your primers. The elongation time is dependent on the DNA that you want to amplify. The amount of cycles required is dependent on the amount of template and the amount of product required. Generally, less cycles is better for dsDNA quality, while more cycles generate more dsDNA fragments.
- Run the PCR product on a 2% agarose gel.
- Sometimes two (or more) bands appear after the PCR. The lowest (smallest) band often corresponds with a PCR product containing only the primers but no DNA of interest. Based on the size of your co-purified ssDNA, you should be able to predict the expected size of a successful generated and amplified dsDNA product (see Figure 2). Cut out the band of the correct size and purify the dsDNA fragments from this band using the GeneJET gel extraction kit according to the protocol provided by the manufacturer.
- If the yield of the dsDNA molecules is too low for cloning/sequencing, the PCR can be repeated on the dsDNA purified from the gel. Ensure to repeat the gel purification, as also products of other lengths might be amplified.
- The purified dsDNA PCR products can be sequenced directly by a modified Illumina sequencing protocol. In order to do this, ensure that primer A contains a sequence suitable for the sequencing platform used (discuss this with your sequencing company). Alternatively, the purified dsDNA PCR products can be cloned in a vector using the introduced KpnI restriction sites on either end of the PCR product (or other restriction sites, if you designed your own primers). Alternatively, the products can be cloned by TOPO cloning (Life Technologies) after addition of 3’-end adenine groups with Taq polymerase (see protocol provided by the manufacturer). After transformation of these vectors, plasmids can be purified and sequenced using standard protocols. After sequencing, the strand originating from the single stranded molecule can be identified by the presence of the 5’-end Primer A sequence and 3’-end poly-A sequence.
- The annealing temperature is dependent on your primers. The elongation time is dependent on the DNA that you want to amplify. The amount of cycles required is dependent on the amount of template and the amount of product required. Generally, less cycles is better for dsDNA quality, while more cycles generate more dsDNA fragments.
- Add 1 µl of DNase free RNase A to 50 µl purified ssDNA.
Representative data
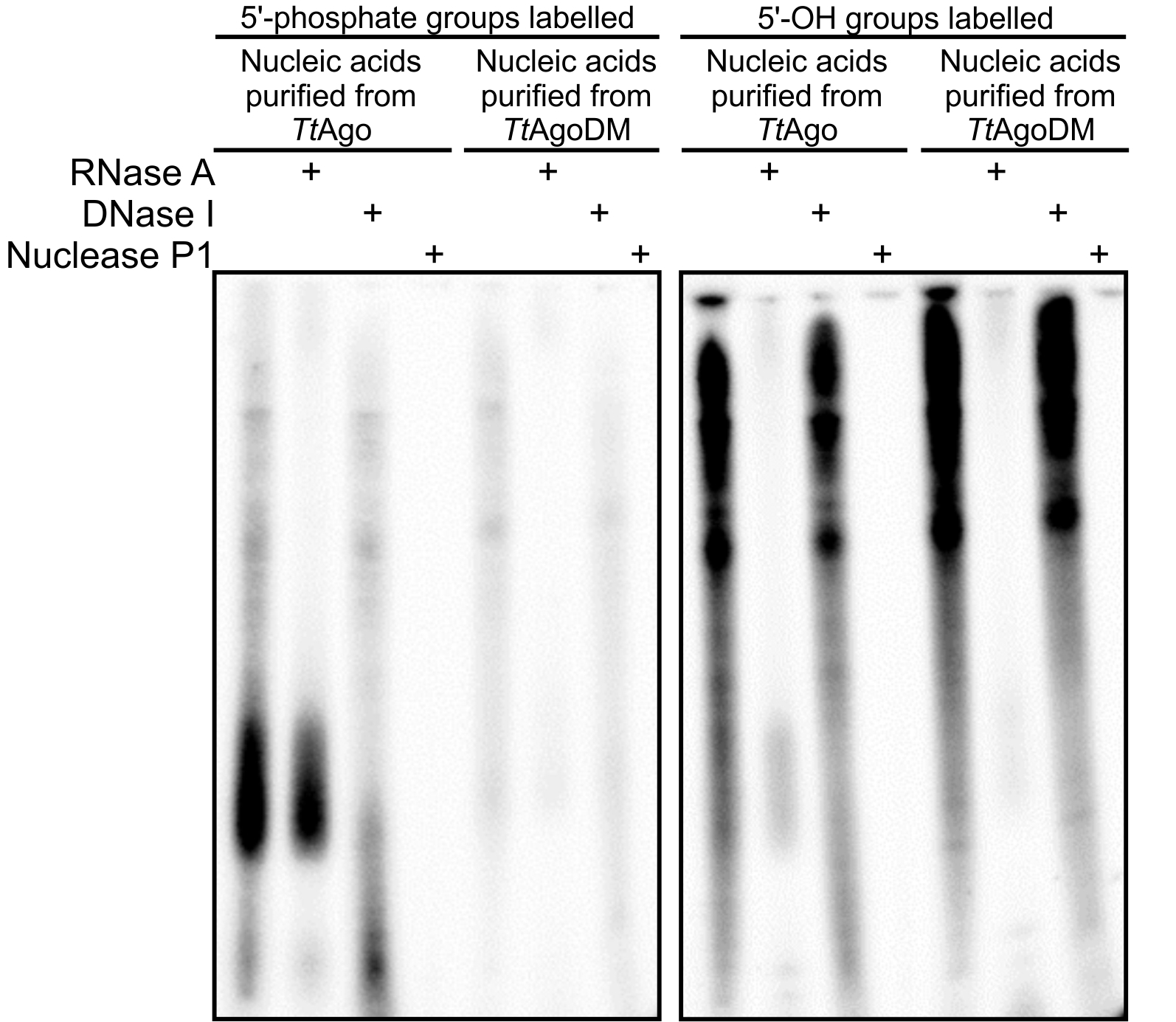
Figure 1. 20% denaturing polyacrylamide gels with resolved nucleic acids that co-purified with TtAgo or TtAgoDM (a catalytic mutant of TtAgo, unable to acquire guides in vivo) (Swarts et al., 2014a). Nucleic acids were 32P labelled by replacing 5’-phosphate (left gel) or 5’-OH groups (right gel) with 32P using T4 polynucleotide kinase. Labelled nucleic acids were cleaved with RNase A, DNase I or nuclease P1 (cleaves ssRNA and ssDNA). The left gel shows that TtAgo co-purified with small ssDNA molecules with a 5’-end phosphate. The right gel shows that both TtAgo and TtAgoDM co-purify with long ssRNA molecules of undefined length. Figure adapted from Swarts et al. (2014a).
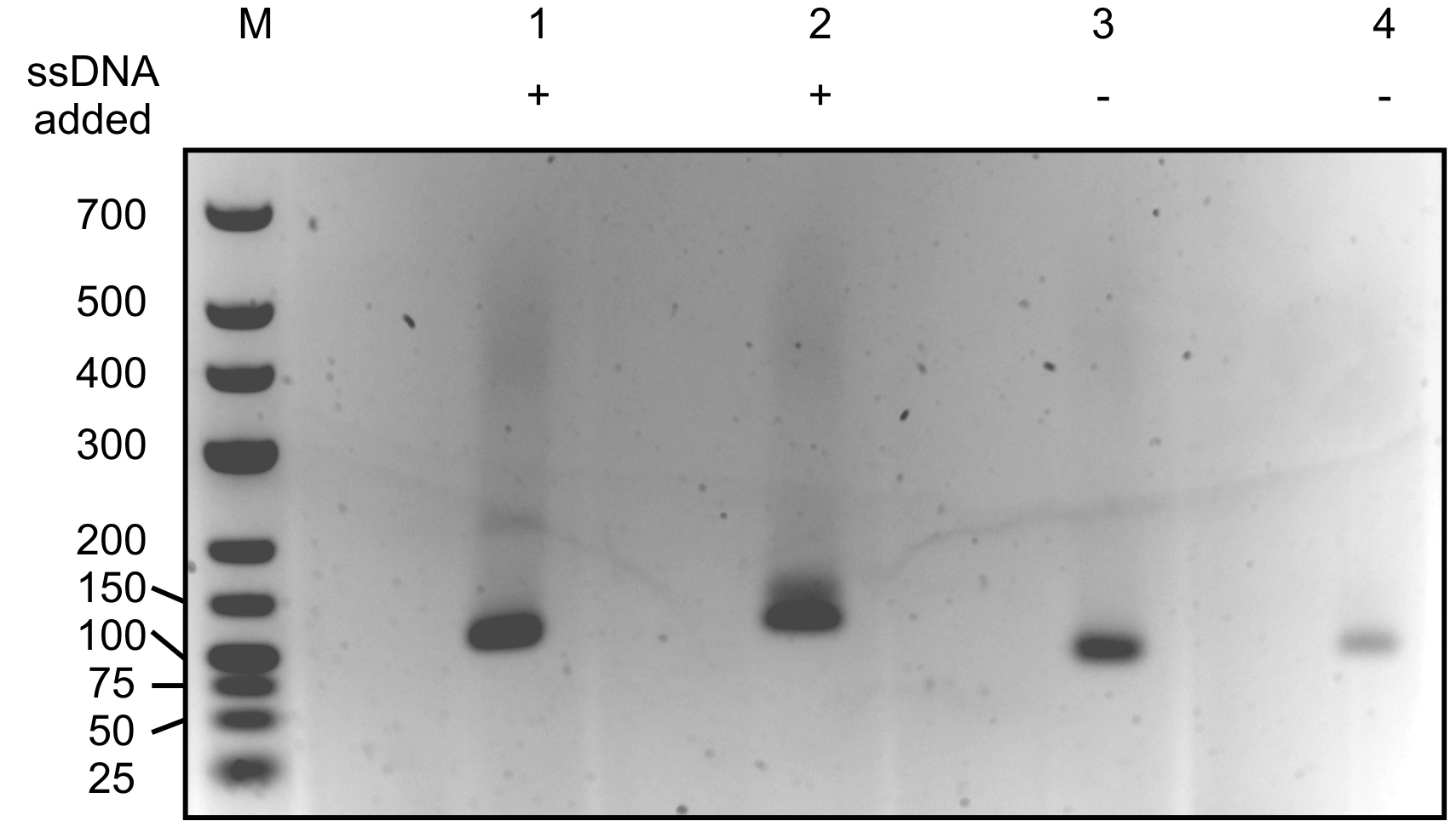
Figure 2. 2% agarose gel with resolved enriched dsDNA molecules. M: Generuler low range DNA ladder. Sizes corresponding with the DNA in the marker lane are indicated in bp. Lanes 1 and 2 contain generated dsDNA samples to which 21 nt ssDNA was added in step B1 of the protocol. Lanes 3 and 4 contain negative controls with generated dsDNA molecules consisting of only the oligonucleotide and 3’-end poly A tail without an ssDNA insert. Note that sometimes, bands representing both molecules with and without an insert will appear in the sample of interest. In this case, make sure you select the right band (usually the upper band, as it contains an insert and thus is larger) for further processing and sequencing.
Acknowledgments
This work was financially supported by a grant from the Netherlands Organization of Scientific Research (NWO) to J.O. (NWO-TOP, 854.10.003).
References
- Swarts, D. C., Jore, M. M., Westra, E. R., Zhu, Y., Janssen, J. H., Snijders, A. P., Wang, Y., Patel, D. J., Berenguer, J., Brouns, S. J. and van der Oost, J. (2014a). DNA-guided DNA interference by a prokaryotic Argonaute. Nature 507(7491): 258-261.
- Swarts, D. C., Jore, M. M. and van der Oost, J. (2014b). Expression and purification of the Thermus thermophilus argonaute protein. Bio-protocol 4(19): e1253.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Swarts, D. C., Westra, E. R., Brouns, S. J. J. and Oost, J. V. D. (2014). Purification and Sequencing of DNA Guides from Prokaryotic Argonaute. Bio-protocol 4(22): e1293. DOI: 10.21769/BioProtoc.1293.
Category
Microbiology > Microbial genetics > DNA
Molecular Biology > DNA > DNA extraction
Molecular Biology > DNA > DNA-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.