- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Purification and Detection of a PDGA Depolymerase from Pusillimonas noertemannii
Published: Vol 4, Iss 21, Nov 5, 2014 DOI: 10.21769/BioProtoc.1280 Views: 9391
Reviewed by: Fanglian HeKanika Gera
Original research article
The authors used this protocol in:

Mar 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The purification of a target protein from a complex mixture of proteins is a challenging undertaking. If the target protein has been previously characterised, then information such as subcellular location, function, molecular weight and pI can be used for the design of a purification strategy. However, if the target protein is uncharacterised or little information regarding its characteristics is available, a generic purification protocol can be employed that is optimised as additional characteristics of the target protein are determined during subsequent purification steps. Herein, we describe the protocol for the purification and detection of a poly-γ-D-glutamic acid (PDGA) depolymerase from a consortium culture of two Gram-negative bacteria using a combination of chromatography, 2D-electrophoresis and zymography.
Materials and Reagents
- Pusillimonas noertemannii BS8 (isolated by soil enrichment)
- Pseudomonas fluorescens BS2 (isolated by soil enrichment)
- Poly-γ-D-glutamic acid >250 kDa [prepared in-house; protocol for production can be found in Negus and Taylor (2014)]
- Protein assay dye reagent (Bio-Rad Laboratories, catalog number: 500-0006 )
- 1.5 ml Microcentrifuge tubes (Eppendorf, catalog number: 0030120086 )
- Laemmli sample buffer (Sigma-Aldrich, catalog number: S3401-10VL )
- IPG strips (immobilized pH 3-10, 7 cm) (Bio-Rad Laboratories, catalog number: 163-2000 )
- Mineral oil (Bio-Rad Laboratories, catalog number: 163-2129 )
- Rehydration trays (Bio-Rad Laboratories, catalog number: 165-4035 )
- Acrylamide solution (Bio-Rad Laboratories, catalog number 161-0156 )
- TEMED (Sigma-Aldrich, catalog number T9281 )
- Agarose overlay solution (Bio-Rad Laboratories, catalog number 163-2111 )
- Zymogram renaturation buffer (Bio-Rad Laboratories, catalog number: 161-0765 )
- Zymogram development buffer (Bio-Rad Laboratories, catalog number: 161-0766 )
- Molecular biology grade water (Merck KGaA, catalog number: H20MB05010 )
- 30% ethanol
- 50% ethanol
- Minimal salts medium broth (see Recipes)
- Anion exchange binding buffer (see Recipes)
- Anion exchange elution buffer (see Recipes)
- Equilibration buffer (see Recipes)
- Non-reducing sample buffer (see Recipes)
- SDS buffer (see Recipes)
- Resolving gel buffer (see Recipes)
- Stacking gel buffer (see Recipes)
- 10% SDS (see Recipes)
- 10% APS (see Recipes)
- 10x TGE running buffer (see Recipes)
- Coomassie-based dye (see Recipes)
- Coomassie destain solution (see Recipes)
- Methylene blue stain (see Recipes)
- 70% ethanol (see Recipes)
Equipment
- Universal tube (Sterilin®, catalog number: 128B/P )
- Erlenmeyer conical flask narrow neck 2 L (Thermo Fisher Scientific, catalog number: 11577422 )
- Cuvettes (1.6 ml semi-micro) (Thermo Fisher Scientific, catalog number: 10594175 )
- Syringe driven filters (0.22 µm, polyethersulfone) (Millex®, catalog number: SLGP033RS )
- Electrode paper wicks (Bio-Rad Laboratories, catalog number: 165-4071 )
- Shaking incubator (Infors AG, model: Multitron standard )
- Spectrophotometer (PerkinElmer, model: Lambda 25 )
- Benchtop centrifuge (Sigma-Aldrich, model: 4-16K )
- Sonicator (MSE, model: Soniprep 150 )
- Exponential sonicator probe (MSE, catalog number: 38121-114A )
- Centrifugal protein concentrators (Sartorious, model: Vivaspin 20, 3 kDa MWCO, catalog number: VS2092 )
- Centrifugal protein concentrators (Sartorious, model: Vivaspin 6, 3 kDa MWCO, catalog number: VS0692 )
- Static incubator (Unitemps, discontinued) (B&T)
- ÄKTAprime plus chromatography system (GE Healthcare, Prime plus, catalog number: 11-0013-13 )
- Anion-exchange chromatography column (GE Healthcare, model: Hi-Trap Q XL, catalog number: 17-5158-01 )
- Orbital rocking platform (Stuart, model: SSM1 )
- Thermal cycler (TECHNE, discontinued, model: FTGENE 2D )
- Electrophoresis cell (Bio-Rad Laboratories, Mini-PROTEAN® Tetra Cell, catalog number: 165-8003 )
- Isoelectric focusing cell (Bio-Rad Laboratories, Protean, catalog number: 165-4000 )
- Vortex mixer (Appleton Woods, Clifton cyclone, catalog number: AA6500 )
Procedure
- Overnight cultures were prepared by rapidly thawing a cryovial containing 1 ml of the depolymerase producing consortium culture and inoculating into 10 ml of minimal salts medium (MSM) broth at a dilution of 1:100 in a 30 ml universal tube. Inoculated cultures were incubated with orbital shaking (120 rpm) at 28 °C for 16 h.
- Overnight cultures were inoculated into 500 ml of MSM broth in a 2 L Erlenmeyer flask at a dilution of 1:100 and incubated with orbital shaking (120 rpm) at 28 °C.
- Optical density readings (OD600) were taken by transferring 1 ml aliquots to cuvettes and reading in a spectrophotometer until OD600 reached 0.5.
- Cells were pelleted by centrifugation at 10,000 x g for 30 min at 4 °C. The cell pellet was resuspended in 10 ml of distilled water and cells disrupted by sonication on ice using an exponential probe (4 x 30 sec bursts at an amplitude of 15 μm at 30 sec intervals).
- Cell debris was removed by centrifugation at 13,000 x g for 10 min at 4 °C and the supernatant passed through a syringe driven filter (0.22 μm pore size) to remove any remaining whole cells.
- The filtrate containing intracellular protein was concentrated approximately 30-fold and submitted to buffer exchange with anion-exchange binding buffer at 4 °C by centrifugation (5,000 x g) in a Vivaspin 20 protein concentrator (3 kDa MWCO).
- Protein concentration of the sample was determined by creating a standard curve using BSA standards at concentrations of 1 mg/ml, 0.5 mg/ml, 0.25 mg/ml and 0.125 mg/ml. Aliquots (20 µl) of protein standard or sample were combined with 1 ml of protein assay dye in a cuvette in duplicate and read at OD595 in a spectrophotometer according to manufacturer’s instruction. The standard curve was used to calculate the concentration of the sample by drawing a line of best fit with linear regression.
- The concentration of the sample was adjusted to 2 mg/ml with anion exchange binding buffer in preparation for anion exchange chromatography.
- A 1 ml Hi-Trap Q XL column was attached to an ÄKTAprime plus fast protein liquid chromatography system and equilibrated at a flow rate of 1 ml/min with 5 column volumes of anion exchange binding buffer, followed by 5 column volumes of anion exchange elution buffer and finally, 10 column volumes of anion exchange binding buffer.
- Intracellular protein (10 mg) in anion exchange binding buffer was bound to the equilibrated column by addition through the injection port of the system in a total volume of 5 ml at a flow rate of 1 ml/min.
- Bound proteins were eluted with a 20 ml linear gradient of NaCl ranging from 0 to 1 M in anion exchange elution buffer at a flow rate of 1 ml/min. Linear gradient is created automatically when using the “ion exchange” application template from the ÄKTAprime menu. The gradient was then held at a NaCl concentration of 1 M for a volume of 17 ml to ensure all bound proteins were eluted.
- Eluting fractions (1 ml/fraction) were collected in 1.5 ml microcentrifuge tubes over the duration of the NaCl gradient and the chromatogram monitored by absorption at 280 nm.
- Individual 1 ml fractions were concentrated approximately 10-fold and subjected to buffer exchange with PBS at 4 °C by centrifugation (5,000 x g) in a Vivaspin 6 protein concentrator. The protein concentration of the concentrated fractions was determined as described in step 7.
- Concentrated fractions were examined for depolymerase activity by incubation with PDGA and analysis of polymer degradation products by SDS-PAGE. Reaction mixtures (20 μl) contained 8 μg of PDGA, 0.5 μg of protein and PBS to volume. Reactions were incubated at 37 °C for 16 h in a static incubator and terminated by combining with combined with an equal volume of 2x Laemmli sample buffer heating at 95 °C for 10 min in a thermal cycler. PDGA degradation products were examined by SDS-PAGE.
- Acrylamide gels were prepared using the Bio-Rad Mini-PROTEAN® Tetra Cell system with a gel thickness of 1 mm using the following formulation:

*Resolving gel buffer: 1.5 M Tris-HCl (pH 8.8)
Stacking gel buffer: 0.5 M Tris-HCl (pH 6.8)
- Samples of reaction mixture (20 µl) were loaded to the gel and electrophoresed at 150 V in SDS buffer until the bromophenol blue dye reached the bottom of the gel.
- Degradation products were visualised by staining gels with methylene blue for 15 min with gentle agitation on an orbital rocking platform. Gels were destained in 30% v/v ethanol (15 min) 30% v/v ethanol (15 min) followed by several changes of distilled water until the background was removed (see Figure 1 for representative result).
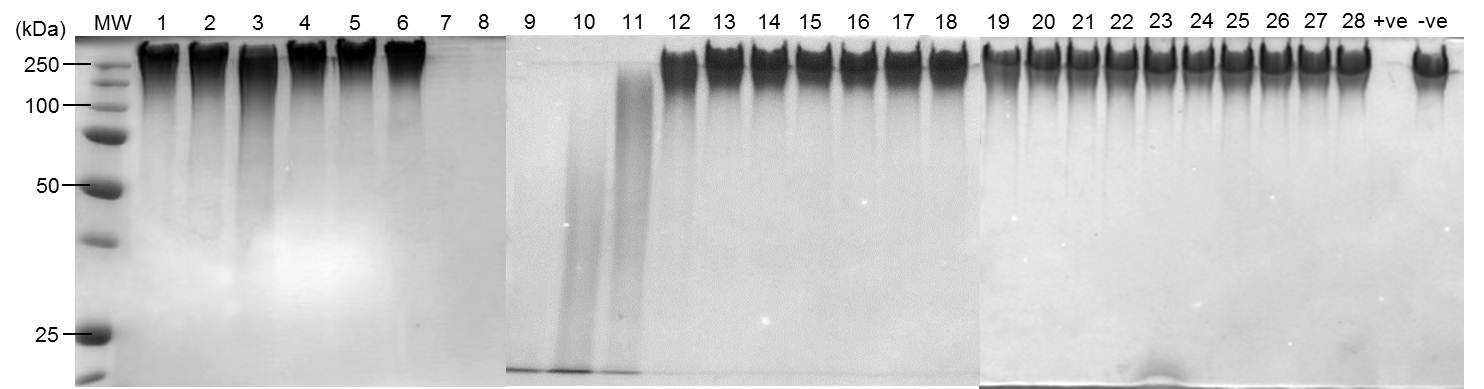
Figure 1. Depolymerase activity of individual fractions separated by anion exchange chromatography. Protein (0.5 µg) from concentrated individual fractions separated by anion exchange chromatography were incubated with PDGA (8 µg) at 37 °C for 16 h in a 20 µl reaction. Degradation products were visualized by SDS-PAGE stained with methylene blue.
- Fractions exhibiting depolymerase activity were pooled and concentrated approximately 30-fold at 4 °C by centrifugation (5,000 x g) in a Vivaspin 6 protein concentrator. The protein concentration of the pooled material was determined as described in step 7.
- For the detection of proteins with depolymerase activity, 2D-zymography was employed. The protein concentration of the partially purified depolymerase preparation (pooled and concentrated fractions) was adjusted to 2 mg/ml with PBS and aliquots containing 20 μg of total protein were combined with 105 μl of non-reducing sample buffer. Samples were mixed vigorously by vortexing and applied immediately with a pipette to a 7 cm IPG strip (pH 3-10) in a rehydration tray for isoelectric focusing.
- To prevent evaporation of the sample, mineral oil (approximately 1 ml) was used to overlay the strips. The rehydration tray was covered with a lid and the strips left to absorb the sample overnight at room temperature.
- Electrode paper wicks were moistened with 8 μl of molecular biology grade water and positioned on the electrodes of the focusing tray.
- IPG strips were removed from the rehydration tray with a pair of forceps, placed on dry tissue paper with the gel side facing up and covered with wet tissue paper for 30 sec in order to remove any excess mineral oil from the surface of the strip.
- IPG strips were then positioned on the focusing tray with the gel side facing down. Approximately 2 ml of mineral oil was layered over the IPG strips, completely covering them to prevent sample evaporation during focusing.
- The focusing tray was positioned in a PROTEAN IEF cell and the cover closed. Strips were then focused using the following conditions:
Conditioning step: 250 V for 15 min, rapid ramp
Linear ramp: 250 V to reach 4,000 V for 2 h
Final focusing: 4,000 V for 10 kVh
Hold step: 500 V indefinitely
N.B: temperature of the focusing cell was maintained at 20 °C
Maximum current: 50 μA/IPG strip
- Strips were immediately removed from the tray after completion of the focusing step, and transferred to dry tissue paper with the gel side facing up. Excess mineral oil was removed from the surface of the gel by covering the strips with wet tissue paper for 30 sec. IPG strips were then transferred to a clean rehydration tray with the gel side facing up for equilibration before focusing in the second dimension.
- IPG strips were covered with 2 ml of equilibration buffer and gently agitated on an orbital shaker for 15 min and the buffer decanted.
- IPG strips were removed from the equilibration tray using forceps and placed into the IPG well on a 10% acrylamide gel copolymerised with PDGA at a final concentration of 0.05% w/v. Gels were prepared as described in step 15 with the addition of lyophilised PDGA to the gel mix.
- Agarose overlay solution was layered into the IPG well to secure the IPG strip in place and was allowed to solidify at room temperature. Gels were placed in a Bio-Rad Mini-PROTEAN® Tetra Cell reservoir filled with TGE running buffer and electrophoresed at 125 V until the bromophenol blue dye reached the bottom of the gel.
- After electrophoresis, duplicate gels were either stained with Coomassie-based dye for the visualisation of protein spots or analysed by zymography.
- For the visualisation of protein spots, gels were covered with Coomassie dye for 1 h with gentle agitation on a rocking platform.
- The stain was decanted and the gel was washed with several changes of destain solution on a rocking platform until the background had been removed.
- Gels developed as zymograms were incubated in Bio-Rad zymogram renaturation buffer for 30 min at room temperature with gentle agitation.
- The buffer was decanted and the gel equilibrated in Bio-Rad zymogram development buffer at room temperature for 30 min with gentle agitation.
- A final incubation was performed in fresh Bio-Rad zymogram development buffer at 37 °C for 16 h.
- The gel was washed with distilled water and stained for detection visualisation of copolymerised PDGA with methylene blue for 15 min.
- The gel was then destained in 30% v/v ethanol (15 min) 50% v/v ethanol (15 min) followed by several changes of distilled water.
- Areas of depolymerase activity were identified as clear spots against a dark background where the enzyme had hydrolysed the copolymerised PDGA substrate (see Figure 2 for representative result).
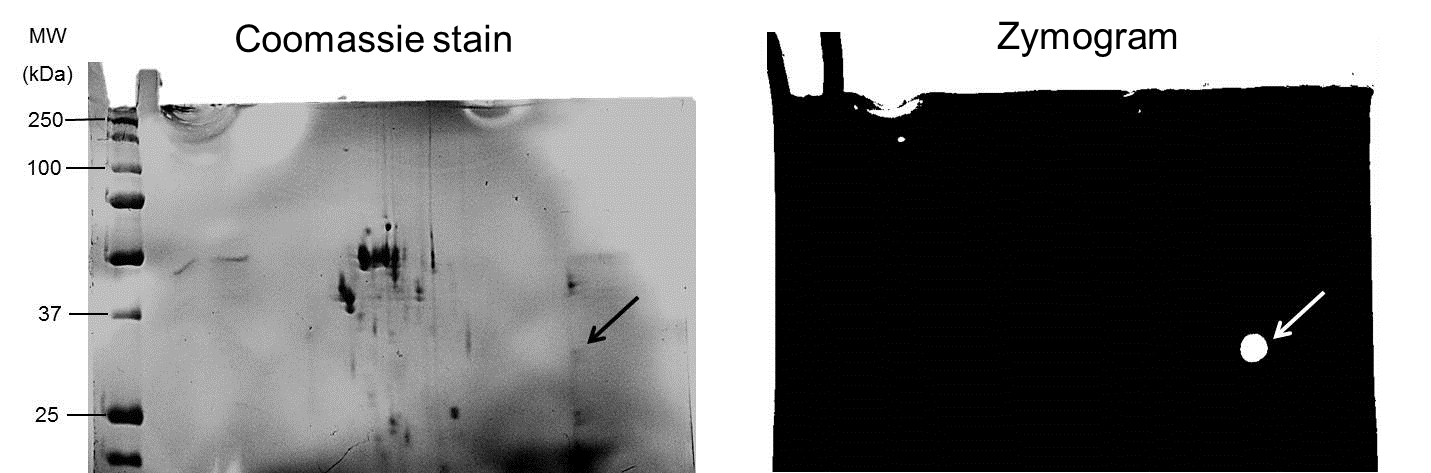
Figure 2. Pooled fractions (20 μg protein) exhibiting depolymerase activity were subjected to 2D-electrophoresis and 2D-zymography in gels containing co-polymerized 0.05% PDGA under non-reducing conditions. In zymograms, depolymerase activity appeared as a bright spot against a dark background (white arrow); corresponding protein spot on Coomassie blue-stained gel indicated by black arrow.
Recipes
- Minimal salts medium broth
4.3 g K2HPO4
3.4 g KH2PO4
2.0 g (NH4)2SO4
0.34 g MgCl2.6H2O
0.026 g CaCl2.2H2O
0.2% w/v PDGA
Distilled H2O to 1,000 ml, sterlise by autoclaving
- Anion exchange binding buffer
20 mM Tris-HCl
Adjust pH to 9.0 with HCl
- Anion exchange elution buffer
1 M NaCl
20 mM Tris-HCl
Adjust to pH 9.0 with HCl
- Equilibration buffer
2% SDS
0.05 M Tris-HCl (pH 8.8)
30% glycerol
Make up to 100 ml dH2O
- Non-reducing sample buffer
4 M urea
4% CHAPS
0.2% carrier ampholytes
0.002% bromophenol blue
- SDS buffer
2% SDS
0.05 M Tris-HCl
30% glycerol
Adjust pH to 8.8 with HCl
- Resolving gel buffer
1.5 M Tris-HCl
Adjust pH to 8.8 with HCl
- Stacking gel buffer
0.5M Tris-HCl
Adjust pH to 6.8 with HCl
- 10% SDS
10 g SDS
Distilled H2O to 1 L
- 10% APS
100 mg APS
1 ml of distilled H2O
- 10x TGE running buffer
30.0 g Tris-HCl
144.0 g glycine
10.0 g SDS
Adjust volume to 1,000 ml with distilled H2O
- Coomassie-based dye
1 g Coomassie brilliant blue
500 ml methanol
100 ml glacial acetic acid
Distilled H2O to 1,000 ml
- Coomassie destain solution
100 ml glacial acetic acid
200 ml methanol
Distilled H2O to 1,000 ml
- Methylene blue stain
2 g methylene blue
0.01 g KOH
220 ml ethanol
Distilled H2O to 1,000 ml
- 70% ethanol
700 ml ethanol
300 ml distilled
Acknowledgments
This work was supported by a Medical Research Council Capacity Building Studentship award.
References
- Negus, D. and Taylor, P. W. (2014). A poly-gamma-(D)-glutamic acid depolymerase that degrades the protective capsule of Bacillus anthracis. Mol Microbiol 91(6): 1136-1147.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Negus, D. and Taylor, P. W. (2014). Purification and Detection of a PDGA Depolymerase from Pusillimonas noertemannii. Bio-protocol 4(21): e1280. DOI: 10.21769/BioProtoc.1280.
Category
Microbiology > Microbial biochemistry > Protein > Isolation and purification
Microbiology > Microbial biochemistry > Protein > Activity
Biochemistry > Protein > Electrophoresis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.