- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Design of a Transcription-based Secretion Activity Reporter (TSAR) for the Type III Secretion Apparatus of Shigella flexneri and Uses Thereof



Published: Vol 4, Iss 20, Oct 20, 2014 DOI: 10.21769/BioProtoc.1270 Views: 11788
Reviewed by: Kanika GeraAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2014
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Many gram-negative bacterial pathogens, including Shigella flexneri, are able to translocate bacterial proteins, dubbed effectors, across the host cell plasma membrane into the host cell cytosol using a syringe-like structure, the type three secretion apparatus (T3SA). While some bacteria use their T3SA to modulate their phagosomal environment (Salmonella spp.), establish pedestal structure to form microcolonies on the plasma membrane (Enteropathogenic Escherichi coli) or lyse their entry vacuole (Shigella spp.), they all have in common a tightly regulated activity of their T3SA. However, the tracking of the activity of the T3SA in infected cells and tissue has been difficult to perform. Using the property of MxiE-dependent promoters that are upregulated when the T3SA is active, we have recently designed a transcription-based secretion activity reporter (TSAR) that allows the following of the activity of the S. flexneri T3SA in real-time in tissue culture cells and in vivo using fast maturing GFP intrinsic fluorescence. Herein we describe the design of the TSAR and its application to fixed and live samples for microscopy and flow cytometry in a colonic epithelial cell model using TC7 tissue culture cells.
Keywords: ShigellaMaterials and Reagents
- Transcription-based secretion activity reporter plasmids (pTSAR1.3, pTSAR1Ud2.1 or pTSAR1Ud2.4s)
Note: Directly available under material transfer agreement (MTA) from Philippe Sansonetti’s laboratory. - Petri dish of Tryptone Casein Soya (TCS) agar (BD Biosciences, catalog number: 236950 ) supplemented with 0.01% Congo red (CR) (SERVA Electrophoresis GmbH, catalog number: 27215.01 ) and the appropriate antibiotic
- TCS Broth (BD Biosciences, catalog number: 211825 )
- Ampicillin (MP Biomedicals, catalog number: 0 219452605 )
- Polylysine (Sigma-Aldrich, catalog number: P1274 )
- Human tissue culture cells such as as colonic epithelial TC7 cells (a clone of Caco-2 cells)
Note: Only polarized epithelial cells permit efficient cell-to-cell spread of Shigella spp. We also recommend using Human cells because it is the sole natural host of Shigella, although most cell lines of other origins tested are also readily infected and could be used for practical reasons. - DMEM (Life Technologies, catalog number: 31885 )
- FCS (Biowest, catalog number: S1810-100 )
- Penicillin/Streptomycin (Life Technologies, catalog number: 15140 )
- Non-essential amino acids (Life Technologies, catalog number: 11140 )
- 0.25% trypsin-EDTA (Life Technologies, catalog number: 25200-056 )
- Fibronectin from human plasma (Sigma-Aldrich, catalog number: F0895 ) (optional)
- HEPES (Life Technologies, catalog number: 15630-056 )
- Gentamicin (Sigma-Aldrich, catalog number: G1397 )
- Cell mask deep red (Life Technologies, catalog number: C10046 )
- Cytochalasin D (Sigma-Aldrich, catalog number: C2873 )
- Live imaging medium containing DMEMgfp-2 (Evrogen, catalog number: MC102)
- PFA (Electron Microscopy Sciences, catalog number: 15714 )
- Glycine (Sigma-Aldrich, catalog number: G7126 )
- Triton X-100 (Sigma-Aldrich, catalog number: T8787 ) (optional)
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A 9647) (optional)
- Gelatin (Sigma-Aldrich, catalog number: G1393 ) (optional)
- Sodium azide (Sigma-Aldrich, catalog number: 0 8591 ) (optional)
- DAPI stock solution (Sigma-Aldrich, catalog number: D9542 ) (optional)
- MOWIOL (Sigma-Aldrich, catalog number: 81381 )
- 1,4-diazobicyclo-[2,2,2]-octaned (DABCO) (Sigma-Aldrich, catalog number: D27802 )
- Glycerol (VWR International, catalog number: 24388.295 ) alternatively Prolong mounting medium (Life Technologies, catalog number: P36930 )
- 10x phosphate-buffer saline (PBS) (see Recipes)
- PBS/10 µg/ml polylysine (see Recipes)
- DMEM-HEPES (see Recipes)
- Chase medium (see Recipes)
- Live imaging medium (see Recipes)
- PBS/4% PFA (see Recipes)
- PBS/100 mM glycine (see Recipes)
- 5,000x DAPI stock solution (see Recipes)
- Mounting medium (see Recipes)
- Growth medium for TC7 cells (see Recipes)
Equipment
- 35 mm petri dish with glass bottom or 8-wells microplate (Ibidi®, catalog numbers: 81158 and 80826 , respectively) for live microscopy
- 24-well plate
- Coverslips no. 1.5, 12 mm in diameter (Harvard Apparatus, catalog number: 64-0712 )
- Laminar flow hood for cell culture
- Heating water bath
- Tabletop centrifuge for 1.5-2 ml Eppendorf tubes
- Tabletop centrifuge with plate-holding rotor
- CO2 incubator for cell culture
- Tweezers
Procedure
- Introductory comments concerning the design of the TSAR
- The design of the TSAR vector was described in detail elsewhere (Campbell-Valois et al., 2014). In brief, expression of a fast maturing variant of GFP (e.g. GFPmut2, GFPmut3 or GFPsfm2) was placed under the control of the promoter of ipaH7.8, a virulence gene regulated by the activity of the Type Three Secretion Apparatus (T3SA). Thus, upon activation of the T3SA, GFP becomes transcribed and translated and allows the monitoring of the T3S system activity (Figure 1).
- Critical elements to optimize the expression level and the signal of the reporter were found to be translation initiation (Shine and Dalgarno sequence and the codon bias of the reporter gene, particularly in the region immediately downstream of the start codon) and the intrinsic properties of the reporter protein used (e.g. brightness, maturation rate etc.). The half-life of GFP, which is normally several hours, could be reduced to around 40 min by fusing the ssrA peptide to the carboxy-terminus, which rendered GFP susceptible to ClpX protease-mediated degradation. Using this approach, we improved the capacity of the TSAR to measure the interruption of the T3SA activity in real-time. Another essential element of the TSAR plasmids is the constitutive expression, using the rpsM promoter, of a second fluorescent protein whose excitation and emission spectra are compatible with the GFP expressed from the T3SA-dependent promoter. The combination of both promoters allowed for following the bacteria in various conditions and assays (microscopy multiple fluorescence, flow cytometer etc.) (Figure 1).
- Using the above mentioned guidelines to optimize the maturation rate, translation initiation, codon usage and half-life as well as selecting the best reporter protein for the desired assay (e.g. RFP, lux operon luciferase, etc.), other reporter systems derived from the original GFP-based TSAR could easily be designed. Here we provide three protocols using the GFP-based TSAR with S. flexneri to assess T3SA activity for fixed samples, live microscopy and flow cytometry.

Figure 1. Plasmid maps. Map of pTSAR1Ud2.1 designed to perform double immunofluorescence followed by microscopic observations A. Map of pTSAR1Ud.4s used in microscopy and particularly live microscopy B. Map of pTSAR1.3 used for FACS applications C.
- The design of the TSAR vector was described in detail elsewhere (Campbell-Valois et al., 2014). In brief, expression of a fast maturing variant of GFP (e.g. GFPmut2, GFPmut3 or GFPsfm2) was placed under the control of the promoter of ipaH7.8, a virulence gene regulated by the activity of the Type Three Secretion Apparatus (T3SA). Thus, upon activation of the T3SA, GFP becomes transcribed and translated and allows the monitoring of the T3S system activity (Figure 1).
- Fixed samples microscopy
- 36-48 h before the experiment, detach a confluent TC7 cells monolayer with 0.25% trypsin/EDTA and distribute TC7 cells on coverslips no. 1.5 placed in wells of a 24-well plate in their normal growth medium. To follow entry events, seed 5 x 104 cells/well (approximately 50-75% confluence the day of the experiment). To favor cell-to-cell spread during longer infection times, seed 1 x 105 cells/well. (Optional) Fibronectin-coated coverslips can be used to permit faster attachment of the cells, but in this case, the number of cells seeded must be reduced about 25-50% to obtain confluence similar to what is described for non-coated coverslips.
- The day before the experiment, a CR–positive colony harboring pTSAR1ud2.1 or pTSAR1Ud2.4s is picked from a CR-containing TCS ampicillin plate and used to inoculate one tube of 8 ml TCS broth supplemented with Amp and incubated overnight at 30 °C with shaking.
Notes:- CR induces T3SA activation and effector secretion; colonies with an active T3SA therefore display a red center where CR has accumulated while bacteria that have lost the virulence plasmid show up as larger white colonies (Figure 2).
- Strains harboring pTSAR1Ud2.1 are used in place of pTSAR1Ud2.4 for multiple labeling experiments because it allows the use of the red laser/Cy3 filter for immunofluorescence.
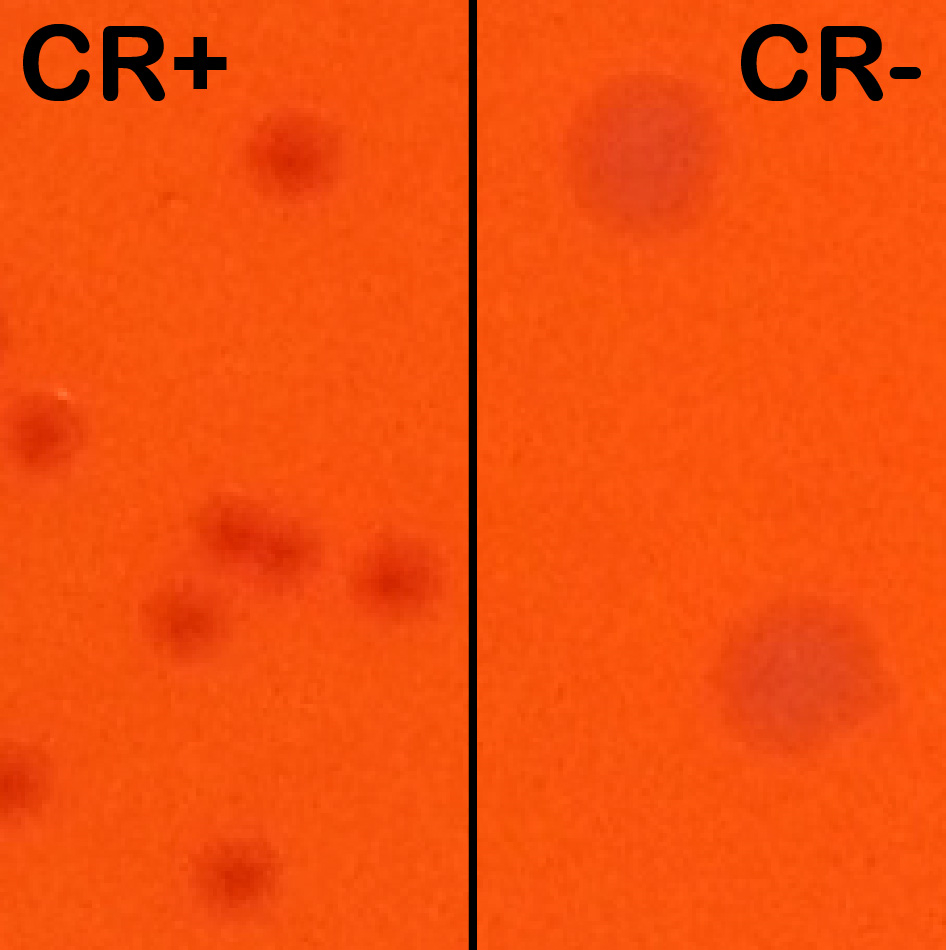
Figure 2. Congo red positive (CR+) versus congo red negative (CR-) colonies. Wt and plasmid cured M90T derived strains were streaked side-by-side on an agar-containing TCS and congo red plate to show their differing phenotypes. Wt colonies on the left are forming small and CR+ colonies (displaying a red center). Plasmid cured (BS176 strain) are forming large CR- colonies (white). - CR induces T3SA activation and effector secretion; colonies with an active T3SA therefore display a red center where CR has accumulated while bacteria that have lost the virulence plasmid show up as larger white colonies (Figure 2).
- The next morning, the ON culture is subcultured 1:100 in a tube of 8.0 ml fresh TCS broth without antibiotics and incubated at 37 °C until the OD600 is approximately 1.0 (late exponential phase).
- 2 ml of bacterial culture (sufficient for the infection of 4 wells of a 24-wells plate) is centrifuged at 7,500 x g for 1 min and washed in 1 ml PBS. Repeat two other times the centrifugation and washing steps.
- Resuspend the bacterial pellet in 2 ml DMEM-HEPES at room temperature (RT).
- TC7 cells are washed 2 times with DMEM-HEPES (RT), after the washes place DMEM-HEPES at 37 °C for step B9.
- 500 µl of the DMEM-HEPES bacterial suspension is added to each well of the 24-well plate seeded with TC7 cells.
- Bacteria are brought into contact with the cells by centrifugation for 5 min at 450 x g at RT. Cells are maintained in contact with the bacterial suspension for approximately 10 min at RT in total. (Optional) if desirable, polylysine-coated bacteria can be used instead of centrifugation. Polylysine-coated bacteria can be prepared by incubating bacteria, which were washed once with 1 ml PBS (see step 4), in PBS/polylysine (10 µg/ml) for 10 min with gentle agitation and washing of the bacteria three times prior to infection. Entry efficiency is greatly enhanced using polylysine treatment, so a bacterial suspension with OD600 equal to 0.02-0.2 should be incubated with the cells at RT for 10 min without centrifugation.
- The bacteria suspension is aspirated and replaced with 500 µl DMEM-HEPES preheated to 37 °C.
- Incubate between 15 to 30 min in a 37 °C cell culture incubator with 5% CO2.
- Aspirate medium and add the chase medium for the desired time. To study cell-to-cell spread in TC7 cells, a chase of 210 min is ideal.
- At the desired time, wash the cell monolayer once with PBS.
- Fix for 10 min with diluted 200 µl PBS/4% PFA at RT.
- Aspirate the fixative and incubate 5 min in 200 µl PBS/glycine to quench the PFA.
- (Optional) DAPI staining (0.2 µg/ml) can be performed in PBS by incubating the cells during 20 min.
- Wash 3 times with PBS.
- (Optional) If desired, cells can be permeabilized to perform immunofluorescence labeling following assay-specific protocols. We routinely use 0.1% Triton X-100 for 10 min to permeabilize cells while maintaining GFP intrinsic fluorescence. Saponin is also usable for this application, while methanol/acetone-based methods are not. Blocking is performed in PBS/1%BSA/0.2% gelatin. Standard immunofluoresence procedure should then be used to stain the coverslip.
Note that in this case, the DAPI staining is performed, as described above, during incubation with the secondary antibodies. - Take out the coverslips from the plate with a fine pair of tweezers, briefly immerse in distilled water and remove excess water using absorbent paper.
- Mount on a slide using 10 µl of pre-warmed (37 °C) mounting medium.
- Let dry without disturbing the CS for at least one hour at room temperature.
- Store at 4 °C until ready to observe.
- The GFP signal in the bacteria is visualized using a GFP or FITC filter or with the 488 nm band of an argon laser. The Cerulean signal is visualized using a CFP filter or with the 458 nm band of an argon laser. mCherry is visualized using a Cy3 filter or with a 568 nm laser. We do not recommend using DsRed in conjunction with GFP in microscopy applications.
- 36-48 h before the experiment, detach a confluent TC7 cells monolayer with 0.25% trypsin/EDTA and distribute TC7 cells on coverslips no. 1.5 placed in wells of a 24-well plate in their normal growth medium. To follow entry events, seed 5 x 104 cells/well (approximately 50-75% confluence the day of the experiment). To favor cell-to-cell spread during longer infection times, seed 1 x 105 cells/well. (Optional) Fibronectin-coated coverslips can be used to permit faster attachment of the cells, but in this case, the number of cells seeded must be reduced about 25-50% to obtain confluence similar to what is described for non-coated coverslips.
- Live microscopy
- TC7 cells are passed at the desired density in regular growth medium 48 h before the experiment in a 35 mm dish with a glass bottom or in an 8-well microplate, if several conditions are to be tested in parallel.
Note: We have also used HeLa and LLC-MK2 cells.
A bacterial suspension of bacteria harboring the pTSAR1Ud2.4s is prepared as above and polylysine coated bacteria are routinely used (see step B8), particularly to monitor entry as entry efficiency is greatly enhanced by this pretreatment. Centrifugation-induced infection without precoating bacteria with polylysine can also be performed by carefully securing the dish or microplate to the plate holder of the centrifuge (see steps B3-8).
Note: Entry of polylysin coated bacteria can be blocked by pretreating cells with 2 µM cytochalasin D, while adhesion to host cells is maintained. Entry can then be triggered within approximately 20 min on HeLa cells by aspirating the cytochalasin D containing media, washing the cell monolayer once and replacing the medium with live imaging medium. This last step can be performed on the microscope stage to allow monitoring of early events leading to entry. - Perform entry and chase of bacterial infection, as described (see steps B9-11), by adjusting incubation time as desired.
- (Optional) The plasma membrane of infected cells can be labeled immediately prior to microscopic observations using Cell Mask Deep Red. The staining is performed by incubating the cells for 5 min with DMEM-HEPES containing 1:1,000 dilution of the Cell Mask. Stained cells are washed three times with DMEM-HEPES and proceed to step C4 just prior to imaging.
- Wash the cells monolayer once with 2 ml live imaging medium or PBS, aspirate, and add 2 ml live-imaging medium to the cell monolayer.
- Transfer the infected cells to the stage of the microscope, which ideally should be equipped with a controlled atmosphere chamber (e.g. 37 °C and 5% CO2).
- To track bacteria, acquire at least 0.5-1 image per minute in fast acquisition mode (using 2 x 2 binning for pixels, for example, can reduce acquisition time). GFP, mCherry and Cell Mask Deep Red can be imaged simultaneously on most microscopic systems used using Cy3 and Cy5 filters, respectively.
- TC7 cells are passed at the desired density in regular growth medium 48 h before the experiment in a 35 mm dish with a glass bottom or in an 8-well microplate, if several conditions are to be tested in parallel.
- Preparation of samples for flow cytometry (FC) analysis
- Pass TC7 cells as above but use one to two wells of a 6-well plate per condition or kinetic point that is to be studied. Distribute 1 x 105-2 x 105 cells per well according to the level of confluence desired for the experiment, taking into account that higher confluence will favor cell-to-cell spread but reduce the initial entry of bacteria.
- Grow bacteria as described above. Strains harboring pTSAR 1.3 are ideal for flow cytometry (FC) because constitutive expression of the DsRed MS165 from the ribosomal rpsM-promoter allows tracking non-secreting bacteria.
Note: In this protocol, we suggest using polylysine-coated S. flexneri to favor entry and maximize recovery of bacteria for FC analyses (see step B8). - TC7 cells are washed two times with DMEM-HEPES (RT).
- 2 ml of the DMEM-HEPES bacterial suspension is added to each well of the 24-wells plate containing TC7 cells.
- Induction of infection is performed as described above (see steps B3-8).
- The infection is allowed to proceed for the desired time (see steps B9-11).
- Bacteria are recovered from the infected cells by detergent lysis, fixation and centrifugation as described in (Aussel et al., 2011). Briefly, cells are washed once in PBS, lysed in 220 µl PBS/0.1% Triton X-100, scraped off the plate, and fixed during 60 min by the addition of 440 µl PBS/3% PFA. Bacteria are then transferred into a 1.5 ml tube and recovered by two centrifugation steps: 5 min at 200 x g to pellet the cellular nuclei; recover the supernatant without disturbing the pellet and centrifuge the supernatant for 10 min at 16,000 x g.
- The pellet is recovered with 50 µl PBS/100 mM glycine.
- After 10 min at RT, samples are further diluted 1:10 in PBS.
- (Optional) samples at this stage can be kept at 4 °C, protected from light, for at least three weeks.
- Samples are transferred to FC tubes and can be further diluted so that approximately 1,000-3,000 events are recorded per second.
- To adjust the compensation and other FC parameters, it is practical to use dilutions of bacterial cultures used for infection and non-labeled and singly labeled strains that were previously fixed with PFA as indicated above.
- Pass TC7 cells as above but use one to two wells of a 6-well plate per condition or kinetic point that is to be studied. Distribute 1 x 105-2 x 105 cells per well according to the level of confluence desired for the experiment, taking into account that higher confluence will favor cell-to-cell spread but reduce the initial entry of bacteria.
Notes
- Results are usually highly reproducible. However, to ensure that the ratio of secreting bacteria to not-secreting bacteria is constant at the given time post-challenge under scrutiny, it is recommended to ensure that the infected cells be studied at a similar level of confluence because when bacteria are unable to perform cell-to-cell spread efficiently, the proportion of immobile, and hence not-secreting bacteria, increases. Similarly, bacteria subculturing prior to infection must be performed in highly controlled conditions, for example, by taking care of stopping incubation at similar OD600 each time an experiment is repeated.
Recipes
- 10x phosphate-buffer saline (PBS)
Mix 80 g of NaCl with 2 g of KCl, 14.4 g of Na2HPO4, 2.4 g of KH2PO4
Adjust pH to 7.4 with NaOH
Add distilled H2O to 1,000 ml
Filter or autoclave
Stored at RT
For all applications, a 1x solution is used - PBS/10 µg/ml polylysine
PBS
1:1,000 of the polylysine stock solution (10 mg/ml in water, filter sterilized and stored at -20 °C) - DMEM-HEPES
DMEM
20 mM HEPES - Chase medium
DMEM
5% FBS
50 µg/ml gentamicin - Live imaging medium
DMEMgfp-2
5% FBS
50 µg/ml gentamicin
Note: Ringer’s buffer can be used in place of the live imaging medium, particularly for infection of short duration. - PBS/4% PFA
10 ml PFA (32%)
8 ml 10x PBS
Add distilled H2O to 80 ml
Aliquot and stored at -20 °C
Discard aliquot after a week at 4 °C - PBS/100 mM Glycine
0.375 g glycine
5 ml 10x PBS
Add H2O to 50 ml
Filter sterilized (0.2 µm)
Stored at 4 °C - 5,000x DAPI stock solution
1 ml H2O
1 mg DAPI - Mounting medium
Note: Recipe from the University of Rochester Medical Center, Michael Mastrangelo, Eric Yehling; for details see http://www.urmc.rochester.edu/confocal-conventional-microscopy/documents/Mowiol52810.pdf.
24 g analytical grade glycerol
9.6 g Mowiol 4-88
24 ml distilled H2O
48 ml 0.2 M Tris buffer (pH 8.5)
To dissolve completely assemble the solution in a 500 ml flask and stir on a heating plate at 60-70 °C for 4-5 h
Aliquot in 20-40 ml per 50 ml conical
Centrifuge at 5,000 x g for 15 min to remove undissolved residues and carefully recover the supernatant
Add 2.5% 1,4-diazobicyclo-[2,2,2]-octane (DABCO) as anti bleaching agent
Aliquot in 15 or 50 ml conicals
Stored at -20 °C for long-term storage - Growth medium for TC7 cells
DMEM
20% FCS
1x Penicillin/Streptomycin
1x Non-essential amino acids
Acknowledgments
We are grateful to Claude Parsot for his comments on the manuscript. FXCV was a CIHR, EMBO, Marie-Curie-IRG and FRM fellow. PS was an EMBO and Marie Curie-IEF fellow while PJS is a HHMI senior scholar. This work was supported by the ERC (PJS Advanced Grant, no. 232798).
References
- Aussel, L., Zhao, W., Hebrard, M., Guilhon, A. A., Viala, J. P., Henri, S., Chasson, L., Gorvel, J. P., Barras, F. and Meresse, S. (2011). Salmonella detoxifying enzymes are sufficient to cope with the host oxidative burst. Mol Microbiol 80(3): 628-640.
- Campbell-Valois, F. X., Schnupf, P., Nigro, G., Sachse, M., Sansonetti, P. J. and Parsot, C. (2014). A fluorescent reporter reveals on/off regulation of the Shigella type III secretion apparatus during entry and cell-to-cell spread. Cell Host Microbe 15(2): 177-189.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Campbell-Valois, F. X., Schnupf, P. and Sansonetti, P. J. (2014). Design of a Transcription-based Secretion Activity Reporter (TSAR) for the Type III Secretion Apparatus of Shigella flexneri and Uses Thereof. Bio-protocol 4(20): e1270. DOI: 10.21769/BioProtoc.1270.
Category
Microbiology > Microbial biochemistry > Protein > Activity
Molecular Biology > Protein > Expression
Cell Biology > Cell imaging > Fixed-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.