- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Genomic Signature of Homologous Recombination Deficiency in Breast and Ovarian Cancers
Published: Vol 3, Iss 13, Jul 5, 2013 DOI: 10.21769/BioProtoc.814 Views: 15354
Reviewed by: Lin Fang
Original research article
The authors used this protocol in:

Nov 2012
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Homologous recombination deficiency, mainly resulted from BRCA1 or BRCA2 inactivation (so called BRCAness), is found in breast and ovarian cancers. Detection of actual inactivation of BRCA1/2 in a tumor is important for patients’ treatment and follow-up as it may help predicting response to DNA damaging agents and give indication Homologous recombination deficiency, mainly resulted from BRCA1 or BRCA2 inactivation (so called BRCAness), is found in breast and ovarian cancers. Detection of actual inactivation of BRCA1/2 in a tumor is important for pat for genetic testing. This protocol describes how to detect impairment of homologous recombination based on the tumor genomic profile measured by SNP-array. The proposed signature of BRCAness is related to the number of large-scale chromosomal breaks in a tumor genome calculated after filtering and smoothing small-scale alterations. The procedure strongly relies on good quality SNP-arrays preprocessed to absolute copy number and allelic content (allele-specific copy number) profiles. This genomic signature of homologous recombination deficiency was shown to be highly reliable in predicting BRCA1/2 inactivation in triple-negative breast carcinoma (97% accuracy; for more details, see Popova et al., 2012) and predictive of survival in ovarian carcinoma (unpublished data). Authors are grateful to Dominique Stoppa-Lyonnet, Anne Vincent-Salomon, Thierry Dubois, and Xavier Sastre-Garau for their contributions. (Patent was deposited: Reference number EP12305648.3, June 7, 2012)
Data and Software
- Data:
- Whole genome SNP-array profile of a tumor. Affymetrix and Illumina are the major platforms providing high quality SNP-array chips and software for primary normalization. Specific protocols are available in manufacturers' websites www.affymetrix.com or www.illumina.com. SNP array profiles have to be further processed to absolute copy number and allelic content profiles by some software for mining SNP array profiles. Good examples of properly processed SNP-array profiles are in Cancer Cell line collection of Sanger Institute.
http://www.sanger.ac.uk/cgi-bin/genetics/CGP/cghviewer/CghHome.cgi - SNP-array like profiles obtained from Next Generation Sequencing (NGS) also could be used in this protocol if they are processed to absolute copy number and allelic content profiles; however, we do not consider it here in details.
- Whole genome SNP-array profile of a tumor. Affymetrix and Illumina are the major platforms providing high quality SNP-array chips and software for primary normalization. Specific protocols are available in manufacturers' websites www.affymetrix.com or www.illumina.com. SNP array profiles have to be further processed to absolute copy number and allelic content profiles by some software for mining SNP array profiles. Good examples of properly processed SNP-array profiles are in Cancer Cell line collection of Sanger Institute.
- Software:
- Software for primary normalization of SNP-arrays: Genotyping Console, ChAS (both www.affymetrix.com), Genome Studio (www.illumina.com), Aroma package (www.aroma-project.org), tQN for quantile normalization of Illumina arrays (Staaf et al., 2008).
- Software for mining SNP array profiles to obtain absolute copy numbers and allelic contents (allele-specific copy numbers), such as GAP (Popova et al., 2009), PICNIC (Greenman et al., 2010), ASCAT (Van Loo et al., 2010), GPHMM (Li et al., 2011), TAPS (Rasmussen et al., 2011), Absolut (Carter et al., 2012), etc.
- The GAP method and further data processing were realized in R environment (www.r-project.org). However, any other language could be used to perform this analysis, including MatLab, Java, C++, etc.
- Software for primary normalization of SNP-arrays: Genotyping Console, ChAS (both www.affymetrix.com), Genome Studio (www.illumina.com), Aroma package (www.aroma-project.org), tQN for quantile normalization of Illumina arrays (Staaf et al., 2008).
Equipment
- Computers (2 GHz, 2 G RAM, Intel Core 2, 40 G HD)
Procedure
- Preprocessing of SNP array data
- Normalize .CEL files by the appropriate software depending on the array platform.
- Export the normalized data: chromosome, position, Log_R_Ratio (Illumina) or Log_2_Ratio (Affymetrix), B allele frequency (BAF, Illumina) or Allelic Difference (AD, Affymetrix) into a text file.
- Process SNP-arrays by (for example) GAP method (Popova et al., 2009) to obtain (Birkbak et al., 2011) estimation of normal contamination and (Carter et al., 2012) absolute copy number and allelic content profiles (Table 1).
Table 1. Segmented tumor genomic profile (a fragment from Affymetrix OncoScan 300K)
* 1 stands for p arm and 1.5 stands for q arm of chromosome 1; pericentric region is indicated in red.Position Start Position End Chromosome* Length SNPs Copy Number Major Allele 59369 115065314 1 13869 2 1 115067829 121049277 1 639 3 2 143701096 144299541 1.5 47 3 2 144337336 144989346 1.5 19 0 0 145008423 148551158 1.5 252 3 2 148565769 152700090 1.5 554 6 5
- Normalize .CEL files by the appropriate software depending on the array platform.
- Quality control
- Quality control of measured SNP-array profile: software for primary data normalization usually provides quality index for each chip; chips indicated to have marginal quality have to be excluded from further analysis; indication of quality cut-offs could be found in corresponding User Guides.
- Contamination of tumor sample by normal stromal cells: sample with more than 65% of predicted normal cells admixture have to be excluded from further analysis (Note: Measured tumor sample usually represents a mixture of tumor and normal cells in different proportions, which results in different contrast in the measured SNP-array profile; 60-70% of normal contamination is at the limit of current recognition techniques).
- Quality control of copy number and allelic content recognition: pattern of copy number alterations (CNAs) in a tumor genome have to be "interpretable", meaning, copy number variation and allelic imbalance profiles have to be consistent. Unfortunately, there is no reliable measure of such consistency developed; we used manual control of recognition based on the GAP plots (Figure 1). GAP plot of a tumor genome is a two dimensional representation of segmented SNP array profiles, where each circle represents a segment (Popova et al., 2009). Clear and regular structure of the GAP plot indicates consistency (Figure 1A), while chaotic structure indicates inconsistency (Figure 1B). Samples with inconsistent profiles have to be excluded from further analysis.
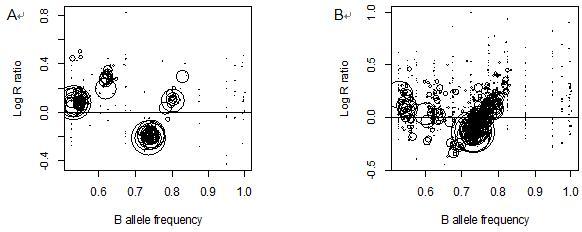
Figure 1. GAP plots for two tumor samples measured by Affymetrix OncoScan 300K representing (A) high quality and (B) low quality profiles. GAP plot of a tumor genome is a two dimensional representation of segmented SNP array profiles, where each circle represents a segment (Popova et al., 2009). Tumor samples are from GEO database (GSE28330, Birkbak et al., 2012). - Quality control of adequate detection of chromosomal breaks: Highly contaminated tumor samples together with unspecific variation in SNP array profiles often result in false positive chromosomal breaks detected by segmentation algorithms; the sample need to be discarded in the case of large number of false positive breaks. Adequate formal procedure for this type of quality control is not yet developed. We performed rough visual estimation of consistency of detected breaks in copy number variation and in recognized copy number profiles.
Note: Poor quality sample comprises around 10-15% of hybridized samples, including low tumor content, poor hybridization, low recognition quality, etc.
- Quality control of measured SNP-array profile: software for primary data normalization usually provides quality index for each chip; chips indicated to have marginal quality have to be excluded from further analysis; indication of quality cut-offs could be found in corresponding User Guides.
- Calculating the number of large-scale chromosomal breaks from segmented profile (Table 1, Figure 2):
Note: Here we describe how to estimate the number of chromosomal breaks related to homologous recombination deficiency; filtering of variation is performed only for the purpose of estimation of breakpoints number and has no relation to particular alterations whatever important they are.- Filtering out micro-variation: The size of micro-variation S_micro is the lower limit of the detectable somatic alteration size, which is dependent on the SNP density in the array; for example, we used 50 SNPs for Affymetrix SNP 6.0; 30 SNPs for Illumina 600K; etc.
Note: Main reason for this filtering is that micro-variations are often linked to germline copy number variations.- Exclude from the segmented genomic profile all segments less than S_micro SNPs and link adjacent segments if they have identical Copy Numbers and Major Alleles.
- Filtering out and smoothing small-scale variation: The size of small-scale variation, S_small < 3 Mb, was defined in (Popova et al., 2012).
Note: Main reason for this definition is that starting from 3 Mb chromosomal breaks follow a Poisson distribution, i.e. are independent from each other; while the small-scale segments tend to cluster in discrete chromosomal regions.- Order small-scale segments according to the size.
- Exclude from the segmented genomic profile the smallest segment and link adjacent segments if they have identical Copy Numbers and Major Alleles.
- Repeat filtering and smoothing until the last small segment.
Note: The way of filtering and smoothing small-scale variations has a minor effect on the resulting profile.
- Order small-scale segments according to the size.
- Calculating number of Large-scale State Transitions (LSTs) of the size S Mb: LST_SMb is defined as a chromosomal break (change in copy number or allelic content) between two adjacent (< 3 Mb in between) segments >= S Mb each; number of LSTs is calculated directly from the segmented genomic profile after filtering and smoothing of small-scale variation (Figure 2).
- Annotate chromosomal breaks as follows: If two segments from the same chromosome arm differ in Copy Number or in Major Allele, and are >= SMb in size, and the distance between the segments is < 3 Mb, the break is annotated as LST_SMb;
- Calculate number of LST_SMb (S = 3, 4,…, 11 Mb) in a tumor genome.
Note: Centromeric breaks are not taken into account.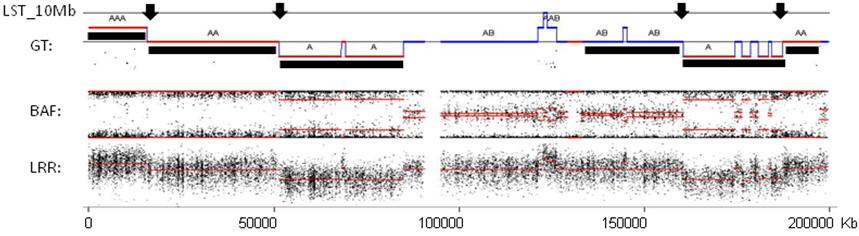
Figure 2. Example of genomic profile of one chromosome with detected copy numbers and LSTs. LRR: log R ratio profile; BAF: B allele frequency profile; GT: segmental genotypes recognized by GAP; LST_10 Mb: black arrows point to LST_10 Mb detected. The black under-line shows large-scale segments obtained after filtering and smoothing small-scale variations seen in the GT profile. Chromosome 3 of a tumor sample from GEO database is shown (GSE28330, Birkbak et al., 2012).
- Annotate chromosomal breaks as follows: If two segments from the same chromosome arm differ in Copy Number or in Major Allele, and are >= SMb in size, and the distance between the segments is < 3 Mb, the break is annotated as LST_SMb;
- Filtering out micro-variation: The size of micro-variation S_micro is the lower limit of the detectable somatic alteration size, which is dependent on the SNP density in the array; for example, we used 50 SNPs for Affymetrix SNP 6.0; 30 SNPs for Illumina 600K; etc.
- Estimation of tumor ploidy:
- Estimate DNA index for a tumor genome as an average copy number in a genome divided by 2.
- Estimate chromosome counts in a tumor genome as a sum of copy numbers at pericentric regions of each chromosome arm (Table 1), following the rules:
- If the size of a segment in pericentric region is >= 1.5 Mb (or 500 SNPs for Affymetrix SNP6.0), the number of copies of corresponding chromosome arm is set to that of the segment;
- If the size of a segment in pericentric region is < 1.5 Mb, chromosome arm count is replaced by its average copy number.
Note: Chromosome number is estimated after filtering micro-variation.
- If the size of a segment in pericentric region is >= 1.5 Mb (or 500 SNPs for Affymetrix SNP6.0), the number of copies of corresponding chromosome arm is set to that of the segment;
- Estimate tumor ploidy following the rule: tumor ploidy is estimated to be 2 (near-diploid genome) if DNA index < 1.3 and chromosome counts < 60; tumor ploidy is estimated to be 4 (near-tetraploid genome) if DNA index >= 1.3 and chromosome counts >= 60.
Note: This attribution is obtained for breast and ovarian cancer genomes based on the analysis of a large number of tumor genomes, (Popova et al., 2012). Genomes with ambiguous attribution of ploidy represented less than 5% of all cases considered. Other cancers might have different genomic evolution and the thresholds for ploidy attribution might need to be adjusted.
- Estimate DNA index for a tumor genome as an average copy number in a genome divided by 2.
- Signature of homologous recombination deficiency in a tumor genome
Based on the analysis of a large series of breast cancers with known status of BRCA1/2 genes the number of LST_6,7,8,9,10 Mb were found to represent effective discriminating features with naturally defined ploidy-specific cutoffs, which allowed prediction of BRCA1/2 inactivation with high accuracy and precision (Table 2). Testing the signature on ovarian cancer showed LST_6,7 Mb to be the most efficient prediction features with similar to breast cancer cohort cut-offs.- Tumor genome is annotated as homologous recombination deficient if number of LSTs in a tumor genome is higher than corresponding ploidy-specific cut-off (Table 2).
Note: Tumors with borderline LST number could be false positives due to false positive breaks detected in the genome. Inconsistency among LST_6, 7, 8, 9, 10 Mb predictions are rare.
Table 2. Cut-offs for LST number predicting BRCAness in breast cancer
*Cut-offs correspond to max (TPR-FPR); cut-offs in parenthesis correspond to 100 sensitivity.LST_S Mb, S Ploidy 2: (p=68, N=182) Ploidy 4: (P=53, N=123) Cut-Off* FPR TPR Cut-Off FPR TPR 6 19 (17) 0.04 0.99 32 (32) 0.10 1 7 17 (15) 0.05 0.99 29 (27) 0.07 0.98 8 14 (14) 0.06 1 26 (26) 0.08 1 9 14 (11) 0.04 0.99 25 (19) 0.07 0.98 10 11 (11) 0.07 1 22 (18) 0.06 0.98
P: Numbers of BRCA1/2 mutated tumors; N: Number of BRCA1/2 wild type or not tested tumors;
TPR: True positive rate; FPR: False positive rate.
- Tumor genome is annotated as homologous recombination deficient if number of LSTs in a tumor genome is higher than corresponding ploidy-specific cut-off (Table 2).
References
- Birkbak, N. J., Wang, Z. C., Kim, J. Y., Eklund, A. C., Li, Q., Tian, R., Bowman-Colin, C., Li, Y., Greene-Colozzi, A., Iglehart, J. D., Tung, N., Ryan, P. D., Garber, J. E., Silver, D. P., Szallasi, Z. and Richardson, A. L. (2012). Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov 2(4): 366-375.
- Carter, S. L., Cibulskis, K., Helman, E., McKenna, A., Shen, H., Zack, T., Laird, P. W., Onofrio, R. C., Winckler, W., Weir, B. A., Beroukhim, R., Pellman, D., Levine, D. A., Lander, E. S., Meyerson, M. and Getz, G. (2012). Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30(5): 413-421.
- Greenman, C. D., Bignell, G., Butler, A., Edkins, S., Hinton, J., Beare, D., Swamy, S., Santarius, T., Chen, L., Widaa, S., Futreal, P. A. and Stratton, M. R. (2010). PICNIC: an algorithm to predict absolute allelic copy number variation with microarray cancer data. Biostatistics 11(1): 164-175.
- Li, A., Liu, Z., Lezon-Geyda, K., Sarkar, S., Lannin, D., Schulz, V., Krop, I., Winer, E., Harris, L. and Tuck, D. (2011). GPHMM: an integrated hidden Markov model for identification of copy number alteration and loss of heterozygosity in complex tumor samples using whole genome SNP arrays. Nucleic Acids Res 39(12): 4928-4941.
- Popova, T., Manie, E., Rieunier, G., Caux-Moncoutier, V., Tirapo, C., Dubois, T., Delattre, O., Sigal-Zafrani, B., Bollet, M., Longy, M., Houdayer, C., Sastre-Garau, X., Vincent-Salomon, A., Stoppa-Lyonnet, D. and Stern, M. H. (2012). Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res 72(21): 5454-5462.
- Popova, T., Manie, E., Stoppa-Lyonnet, D., Rigaill, G., Barillot, E. and Stern, M. H. (2009). Genome Alteration Print (GAP): a tool to visualize and mine complex cancer genomic profiles obtained by SNP arrays. Genome Biol 10(11): R128.
- Rasmussen, M., Sundstrom, M., Goransson Kultima, H., Botling, J., Micke, P., Birgisson, H., Glimelius, B. and Isaksson, A. (2011). Allele-specific copy number analysis of tumor samples with aneuploidy and tumor heterogeneity. Genome Biol 12(10): R108.
- Staaf, J., Vallon-Christersson, J., Lindgren, D., Juliusson, G., Rosenquist, R., Hoglund, M., Borg, A. and Ringner, M. (2008). Normalization of Illumina Infinium whole-genome SNP data improves copy number estimates and allelic intensity ratios. BMC Bioinformatics 9: 409.
- Van Loo, P., Nordgard, S. H., Lingjaerde, O. C., Russnes, H. G., Rye, I. H., Sun, W., Weigman, V. J., Marynen, P., Zetterberg, A., Naume, B., Perou, C. M., Borresen-Dale, A. L. and Kristensen, V. N. (2010). Allele-specific copy number analysis of tumors. Proc Natl Acad Sci U S A 107(39): 16910-16915.
Article Information
Copyright
© 2013 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Popova, T., Manié, E. and Stern, M. (2013). Genomic Signature of Homologous Recombination Deficiency in Breast and Ovarian Cancers. Bio-protocol 3(13): e814. DOI: 10.21769/BioProtoc.814.
Category
Cancer Biology > General technique > Genetics > Genome-wide analysis
Cancer Biology > Genome instability & mutation > Biochemical assays > DNA structure and alterations
Molecular Biology > DNA > DNA recombination
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.