- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Detection and Cloning of Spliced Transcripts by RT-PCR


Published: Vol 3, Iss 8, Apr 20, 2013 DOI: 10.21769/BioProtoc.486 Views: 14663
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2012
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Using a Reverse Transcriptase-PCR approach spliced transcripts can be converted to cDNA, amplified and cloned into an expression plasmid. Sequencing of the obtained cDNA allows identification of the splicing events that generated the detected RNA (Grewe et al., 2012).
Materials and Reagents
I. RT-PCR
- QuantiTect Probe RT-PCR Kit (QIAGEN, catalog number: 204443 )
- Primers (sense and antisense) (Biomers)
- Isolated RNA
- PCR tubes (Starlab)
- Nuclease-free water (B Braun)
II. Cloning
- Agarose (Roth, catalog number: 2267.4 )
- Ethidium bromide (AppliChem)
- DNA ladders (1 kb and 100 bp) (Life Technologies, InvitrogenTM)
- Geneclean III Kit (Qbiogene, catalog number: 1001-600 )
- Example of an acceptor plasmid: pcDNA3.1 (Life Technologies, InvitrogenTM, catalog number: V790-20 )
- Restriction endonucleases with reaction buffers (New England Biolabs)
- Antarctic phosphatase with reaction buffer (New England Biolabs, catalog number: M0289L )
- TaKaRa-DNA-Ligation-Kit version 2.1 (TaKaRa, catalog number: 6022 )
- Ultracompetent XL2-blue bacteria (Stratagene, catalog number: 200150 )
- Carbenicillin (AppliChem, catalog number: A1491 )
- Bacteria cell culture tubes (Sarstedt)
- Plasmid preparation Kits (QIAGEN, catalog number: 27104 , 12362 )
- Tris-HCl (pH 7.8)
- Sodium acetate
- EDTA
- NaCl
- KCl
- MgCl2
- MgSO4
- LB medium (see Recipes)
- TAE buffer (see Recipes)
- 6x loading dye (see Recipes)
- SOC medium (see Recipes)
- LB agar (see Recipes)
Equipment
- PCR cycler (Mastercycler gradient 5331, Eppendorf, catalog number: 5331000.010 )
- Microwave oven (Alaska, model: M 1001 )
- Gel electrophoresis chamber (Perfect Blue electrophoresis system 40-1214, PeqLab) and power supply (Power supply E844, Consort)
- UV light box (UV-Transilluminator) (Vilber)
- Incubators (Thermomixer comfort, Eppendorf, catalog number: 5355000.011 ; 37 °C shaker for bacterial cell cultures Novotron, Infors HT)
- Scalpel (B Braun)
- Scalpel (B Braun)
- Bacteria cell culture tubes (Sarstedt)
Procedure
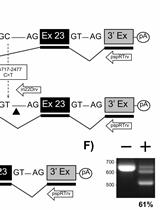
Depending on the experiment isolation of cytoplasmic or total RNA should be done. In general, cytoplasmic RNA is enriched in spliced RNA lacking all introns whereas total RNA contains unspliced, partially-spliced and fully-spliced RNA. A protocol for the isolation of cytoplasmic RNA from mammalian cells can be obtained from the first part of the bio-protocol "Packaging of retroviral RNA into viral particles analyzed by quantitative Reverse Transcriptase-PCR". Sense and antisense primers should hybridize upstream of the splice donor and downstream of the splice acceptor sequence, respectively (Figure 1A). It is also possible that one primer overlaps the exon-exon junction ensuring perfect hybridization to spliced RNA only. For subsequent cloning, both primers should contain recognition sites for restriction endonucleases (cloning via restriction enzymes) or sequences identical to the acceptor plasmid (cloning via homologous sequences) at their 5' ends. A cDNA containing the whole open reading frame (ORF) of a gene can be generated when sense and antisense primers hybridize to the beginning of the ORF sequence or to the 5' untranslated region and to the end of the ORF sequence or the 3' untranslated region, respectively (Figure 1B).
- Set up RT-PCR reaction with the QuantiTect Probe RT-PCR Kit (QIAGEN) as follows:
Also prepare a control without addition of the Reverse Transcriptase (RT) in order to control for (genomic or plasmid) DNA contamination.PCR mix (2x, contains Taq, dNTPs and buffer) 10 μl Reverse Transcriptase enzyme 0.2 μl Primer sense (10 μM) 0.4 μl Primer antisense (10 μM) 0.4 μl Isolated RNA 0.5 to 1 μg template RNA in up to 5 μl Add nuclease-free water to 20 μl.
When the cDNA obtained in the RT-PCR will be inserted in an acceptor plasmid via restriction enzyme digestion it must carry the desired restriction enzyme recognition sites which can be added to the 5' ends of the primers. Be aware that approximately 4 additional random nucleotides should be added upstream of the recognition sites in the primers to allow efficient cleavage to take place. - RT-PCR reaction:
Note: The QuantiTect Probe RT-PCR Kit (Qiagen) allows the RT reaction and the PCR to be performed in one run without interruption.
RT step at 50 °C for 30 min followed by 15 min of 95 °C. Subsequently, amplify the cDNA by 30 PCR cycles starting at 95 °C for 30 sec, 50 to 60 °C depending on the melting temperature of the primers for 30 sec and 72 °C for 30 sec per 500 bp of amplicon length. It is possible to add a final extension step at 72 °C for up to 7 min to allow the DNA polymerase to elongate remaining single stranded DNA. Keep the PCR at 4 °C until agarose gel electrophoresis.
Short elongation steps are necessary to preferentially detect spliced versus unspliced/partially-spliced transcripts. Sometimes it may be necessary to perform a nested-PCR to preamplify the cDNA. Perform the first RT-PCR with primers surrounding the sequence that is amplified in the second PCR. Primers for the second PCR must bind the fragment amplified in the first RT-PCR. One microliter directly from the first RT-PCR (without any purification) should be sufficient as template for the second PCR. However, the first amplicon can also be purified by a PCR clean-up kit and up to 200 ng of DNA per 20 μl reaction can be used as template for the second PCR which can be performed with the QuantiTect Probe RT-PCR Kit omitting the RT step and starting with 15 min at 95 °C. - Prepare 1.5 % agarose gels in TAE buffer containing ethidium bromide (see recipes).
- Add 4 μl of 6x loading dye to the 20 μl PCR reaction and transfer the solution to the agarose gel.
- Agarose gel electrophoresis is done for approximately 30 min at 100 to 120 V (5 to 6 V per cm distance between electrodes). Analyze DNA ladder(s) in parallel to be able to determine the DNA fragment sizes. Please note that Ethidium bromide molecules are positively charged and move in the opposite direction in the agarose gel as the DNA molecules. Analyze the gel when the loading dye band reaches approximately the middle of the agarose gel (see Figure 1D).
- Document the result under a UV lamp gel documentation system. The intercalated Ethidium bromide allows detection of the amplified cDNA fragments (Figure 1D). Since UV light can damage the DNA, reduce the exposure time to a minimum.
- Cut out small gel pieces containing the desired DNA fragments at the UV table with a clean scalpel. Please wear laboratory safety glasses and reduce the exposure time to the UV light to a minimum.
- Extract the DNA from the gel pieces with the Geneclean III Kit. Elute the DNA from the glass beads with 10 μl of nuclease-free water.
- Prepare a restriction enzyme digestion of the isolated cDNA (the entire 10 μl) and of approximately 3 μg acceptor plasmid DNA in parallel:
Incubate for 1 to 3 h at 37 °C. When more than one restriction enzyme is used or when restriction enzymes are used that have star activity please increase the reaction volume to 30 μl. The enzyme(s) should not constitute more than 10 % (v/v) of the reaction volume. Star activity can also be reduced by decreasing the digestion time. However, complete digestion is critical for successful ligation and transformation. Extended incubation times up to overnight should only be performed with restriction enzymes without star activity or in increased reaction volumes (up to 50 μl).DNA solution 10 μl Restriction buffer (10x) 2 μl Bovine serum albumin solution (10x) 2 μl Restriction enzyme(s) (20 U/μl) 1 μl Add water to 20 μl
When compatible cohesive ends are generated during digestion (e.g. by using just one restriction enzyme) the linearized acceptor plasmid must be dephosphorylated to prevent self-ligation. After restriction enzyme digestion add Antarctic phosphatase together with its buffer directly to the reaction mix. After 30 min at 37 °C the enzyme should be inactivated by incubation for 5 min at 65 °C followed by agarose gel electrophoresis.
For transient expression of genes in mammalian cells the plasmid pcDNA3.1 (Life Technologies, InvitrogenTM) (http://tools.invitrogen.com/content/sfs/vectors/pcdna3.1+.pdf) can be used as an acceptor plasmid. Due to the immediate early Cytomegalovirus (CMV) enhancer/promoter and the Bovine Growth Hormone (BGH) polyadenylation sequence it allows high expression levels. The cDNA from the RT-PCR must be inserted into the multiple cloning site. In addition, an efficient Kozak sequence (A/GCCATGG) should be added surrounding the start codon of the open reading frame which can be implemented into the sense primer during RT-PCR. - Repeat steps 3 to 8 with the digested DNA. Cut out small gel slices containing only the linearized acceptor plasmid. Contamination with still circular plasmid DNA will lead to efficient re-transformation of the original plasmid and strongly reduce the amount of bacterial colonies carrying the insert-containing plasmid.
- Mix 2 μl of the digested and purified insert, 3 μl of the digested and purified acceptor plasmid DNA and 5 μl of solution I of the Takara Ligation Kit. Incubate at 16 °C for overnight. Alternatively, the DNA concentration can be measured and a vector: Insert DNA ratio of 0.03 pmol: 0.03 - 0.3 pmol can be used for ligation (manufacturer's instructions from the TaKaRa-DNA-Ligation-Kit version 2.1).
A detailed protocol for transformation of DNA and subsequent amplification of plasmids in bacteria can be found in the bio-protocol "Standard DNA Cloning" (He, 2011b) and "Plasmid DNA Extraction from E. coli Using Alkaline Lysis Method" (He, 2011a) (please generate hyperlink).
In brief, the following steps have to be performed: - Transform ultracompetent XL-2 bacteria with 1.5 μl of the ligation reaction by heat shock.
- Let transformed bacteria grow for 1 h at 37 °C in SOC medium without antibiotics.
- Cultivate bacteria on LB agar plates containing an appropriate antibiotic at 30 to 37 °C overnight.
- Pick single colonies and let them growth overnight at 30 to 37 °C in 3 to 6 ml LB medium containing an appropriate antibiotic.
- Extract plasmid DNA from 1.5 to 3 ml of the bacterial culture. Keep the rest of the cultures at 4 °C.
- Digest the plasmid DNA with appropriate restriction enzymes to identify bacteria colonies containing successfully generated plasmids. In addition, sequence those plasmids that show fitting restriction enzyme digestion patterns. This sequencing reaction allows identifying the splicing event that actually generated the detected and cloned RNA.
- Prepare Midi, Maxi or Giga plasmid preparations from the identified bacteria colonies harboring the successfully generated plasmid. Endotoxin-free plasmid preparation kits avoid the presence of lipopolysaccharide in the plasmid DNA solution that will be used for transfections or to treat animals.

Figure 1. Principle of the RT-PCR approach for the detection and cloning of spliced RNA. A. Sense and antisense primers (horizontal arrows) hybridize adjacent to the exon-exon boundary generated by fusion of splice donor 1 (SD1) and splice acceptor 1 (SA1) sequences during splicing. Restriction enzyme recognition sites (RE1 and RE2, vertical arrows) are present in the plasmid DNA and in the RT-PCR-amplified DNA generated from the partially-spliced RNA which can be used to insert the PCR fragment into the plasmid. B. Sense and antisense primers (horizontal arrows) contain restriction enzyme recognition sites (RE1 and RE2, vertical arrows) at their 5' ends. They hybridize in the 5' and 3' untranslated regions (UTR). Amplification of the fully-spliced RNA by RT-PCR allows insertion of the DNA into a plasmid (e.g. pcDNA3.1) using restriction enzymes 1 and 2. C. Depicted is a plasmid used to transfect HEK293T cells and its transcripts. RNAs generated by splicing between SD1 and SA5 or after an additional splicing event between SD4 and SA7 were reinserted into VHgenomic generating the plasmids VHenv or VHnef, respectively (Grewe et al., 2012). D. Shown is the agarose gel electrophoresis result obtained after RT-PCR amplification of spliced RNA from RNA isolated from cells transfected with the plasmid VHgenomic using primer set 1 containing forward primer and antisense primer 1 or primer set 2 containing forward primer and antisense primer 2 (see horizontal arrows in C) (Grewe et al., 2012). SD1-SA5 or SD1-SA5, SD4-SA7 indicates the splicing events detected after sequencing of the cDNA.
UTR, untranslated region; RE, restriction enzyme recognition site; white bars, introns; dark grey bars, open reading frame; polyA, polyadenylation signal; SD, splice donor; SA, splice acceptor; horizontal arrows, primers; vertical arrows, location of restriction enzyme recognition sites; black dot, RNA 5' CAP; black oval; 3' polyA tail; LTR, lentiviral long terminal repeats; CMV, cytomegalovirus immediate early enhancer/promoter; GFP, green fluorescence protein.
Recipes
- 1x TAE buffer
40 mM Tris-HCl (pH 7.8)
5 mM Sodium acetate
1 mM EDTA - Agarose
1.5 % (w/v) agarose in 1x TAE buffer
Cook for 1 min in microwave oven and shake carefully
Add 0.7 μg/ml Ethidium bromide [e.g. 7 µl of 1 % ethidium bromide solution (10 mg/ml) per 100 ml] after the solution is cooled down but still liquid and shake carefully. - 6x loading dye
30 mM Tris-HCl (pH 7.6)
1 mM EDTA
35 % (v/v) glycerine
0.35 % (w/v) bromphenol blue - SOC medium
2 % (w/v) tryptone
0.5 % (w/v) yeast extract
20 mM glucose
10 mM NaCl
2.5 mM KCl
10 mM MgCl2
10 mM MgSO4
Sterilize by autoclaving. - LB agar
1.5 % (w/v) Bacto-Agar
In LB-Medium
Sterilize by autoclaving. - LB medium
1 % (w/v) tryptone
0.5 % (w/v) yeast extract
171 mM NaCl
pH 7.4
Sterilize by autoclaving.
Acknowledgments
The protocol was adapted from our paper Grewe et al. (2012). This work was funded by a grant from the German Research Foundation (DFG) to Klaus Überla (Ue45/11-1). Bianca Hoffmann is and Bastian Grewe was supported by a fellowship from the DFG graduate school (GRK 1045). Beside Bianca Hoffmann and Bastian Grewe, Katrin Ehrhardt, Maik Blissenbach, Sabine Brandt, Klaus Überla, Alexander Stang, Thomas Grunwald, Klaus Sure, and Bettina Tippler were part of the team which established the methods described.
References
- Grewe, B., Ehrhardt, K., Hoffmann, B., Blissenbach, M., Brandt, S., and Uberla, K. (2012). The HIV-1 Rev protein enhances encapsidation of unspliced and spliced, RRE-containing lentiviral vector RNA. PloS One 7(11), e48688.
- He F. (2011a). Plasmid DNA extraction from E. coli using alkaline lysis method. Bio-protocol 1(3): e30.
- He F. (2011b). Standard DNA cloning. Bio-protocol 1(7): e52.
Article Information
Copyright
© 2013 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Hoffmann, B. and Grewe, B. (2013). Detection and Cloning of Spliced Transcripts by RT-PCR. Bio-protocol 3(8): e486. DOI: 10.21769/BioProtoc.486.
Category
Molecular Biology > RNA > RNA splicing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.