- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Determination of Toxoplasma gondii Replication in Naïve and Activated Macrophages
(*contributed equally to this work) Published: Vol 2, Iss 22, Nov 20, 2012 DOI: 10.21769/BioProtoc.289 Views: 13310
Original research article
The authors used this protocol in:

Apr 2012
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Toxoplasma gondii is an obligate intracellular protozoan parasite that causes the disease toxoplasmosis. Chronic infection is established through the formation of tissue cysts predominantly in cardiac and neurologic tissues. A defining characteristic of T. gondii is its ability to evade the host’s immune defenses; specifically, T. gondii can invade and persist within host phagocytes, using them to disseminate to the brain and central nervous system where cysts are then formed. This protocol is used to evaluate the ability of Toxoplasma gondii to survive and replicate within naive and activated murine bone marrow-derived macrophages at the level of single infected cells. In the following protocol macrophages are naive or activated with IFN-γ and LPS but different activation stimuli can be utilized as well as different host cell populations and diverse inhibitors. Parasite replication is determined by evaluating the number of parasites per vacuole over time using immunofluorescence staining for parasties and microscopic analysis. Kinetic determination of parasite number per vacuole accurately reflects parasite replication over time as vacuoles-containing parasites do not fuse with one another. Isolation of murine bone marrow-derived macrophages, preparation of conditioned L929 cells for collection of macrophage colony-stimulating factor, and staining for fluorescence microscopy included in the protocol has broad applicability. This protocol works well for pathogens like Toxoplasma gondii that reside in vacuoles that do not fuse with one another and that can be visualized by microscopy.
Keywords: ToxoplasmaMaterials and Reagents
Note: source of reagents is not critical to experiment.
- Murine bone marrow derived macrophages (see protocol below)
- Rabbit polyclonal antibody against Toxoplasma gondii (Fitzgerald Laboratories)
- Alexa Fluor® 488-conjugated goat anti-rabbit IgG (H + L) (Life Technologies, Invitrogen™, catalog number: A-11012 )
- Dulbecco’s modified ragle medium high glucose (DMEM) (1x) (Life Technologies, Invitrogen™, catalog number: 10313-039 )
- Fetal calf serum (FCS) (Hyclone, catalog number: SH30396.03 ); heat inactivated 56 °C for 30 min
- L-gluatamine (Hyclone, catalog number: SH30034.01 )
- Penicillin/streptomycin (Hyclone, catalog number: SV30010 )
- L929 cell (ATCC CCL1) conditioned media as a source of macrophage colony-stimulating factor (M-CSF) for maturation of bone marrow-derived macrophages (see protocol below)
- Dulbeccos PBS (Ca2+ free and Mg2+ free) (DPBS) (Hyclone, catalog number: SH30028.02 )
- Toxoplasma gondii parasites (tachyzoites), Tachyzoites are the haploid replicating stage of Toxoplasma gondii that can be grown in vitro in human fibroblast cells or other host cell types.
- Lipopolysaccharide (LPS) (List Biological from E. Coli O55: B5 #203)
- Recombinant murine IFN-γ (Pepro Tech, catalog number: 50813665 )
- Aminoguanidine (Thermo Fisher Scientific, Acros Organics, catalog number: AC40078-1000 ) - inhibitor of inducible nitric oxide synthase
- Sodium nitroprusside (SNP) (Thermo Fisher Scientific, catalog number: AC211640000 ) - nitric oxide donor
- 16% methanol-free paraformaldehyde EM grade (Electron Microscopy Sciences, catalog number: 15710 )
- Triton X100 (Sigma-Aldrich, catalog number: T9284-500 )
- Mounting media such as Vectashield containing DAPI to stain nucleus (Vector Laboratories, catalog number: H-1200 )
- Cover slips for the chamber slides (22 x 50 x 1) (Thermo Fisher Scientific, catalog number: 12-548-5E )
- 4% paraformaldehyde
- KCl
- KH2PO4
- NaCl
- Na2HPO4
- DPBS (we purchase it but the recipe is below) (see Recipes)
- D10 media (see Recipes)
- BMC media (see Recipes)
- Blocking buffer (see Recipes)
- Wash buffer (see Recipes)
Equipment
- Fluorescence microscope with 100x phase or DIC objective
- Tabletop centrifuge capable of holding 15 or 50 ml centrifuge tubes
- Humidified CO2 incubator at 37 °C
- Bacteriological petri plates (100 mm x 15 mm) (Thermo Fisher Scientific, catalog number: 0875712 )
- Zeiss inverted Axiovert 200
- Hemocytometer
- 8-well chamber slides (BD Biosciences, catalog number: 354118 ) or cover slips inserted into wells in a 24 well plate
- Sterile plastic 50 ml centrifuge tubes (brand is not important)
Procedure
- Isolate murine bone marrow derived-macrophages (see protocol below).
- Grow macrophages for 1 week in BMC media (see recipe and protocol below) on bacteriological petri plates.
- Dislodge macrophages from bacteriological petri plates with cold DPBS (calcium and magnesium free). This is done by rinsing with cold DPBS, discarding the added DPBS, repeating the wash and then keeping the plates with PBS at 4 °C for 20 min and collecting adherent cells by rinsing monolayer with DPBS using a sterile transfer pipette or serological pipette (see detailed protocol below).
- Add macrophages at a concentration of 3 x 105 macrophages per ml in 250-300 μl of D10 medium to chambers at least 2 h before addition of parasites to allow macrophages to adhere. Place cover back on chamber slide and place in humidified chamber at 5% CO2 at 37 °C. Add 1 x 104 Toxoplasma gondii parasites to macrophages. Parasites can be added directly to the 250-300 μl of media already in chambers. Replace slide cover and allow parasites to invade for 4 h in a humidified incubator with 5% CO2 at a temperature of 37 °C. Generally 1-10 μl of parasites adjusted to the appropriate concentration in D10 medium is added per chamber. If you need to adjust the concentration of parasites by centrifugation followed by resuspension in fresh D10 media before adding to chambers use polystyrene or PET (polyethylene terephthalate) centrifuge tubes as parasites stick to polypropylene; centrifugation 1,700 rpm [582 rcf, 10 min].
- Remove the media and rinse the chamber slides with fresh D10 medium to remove any remaining extracellular parasites. In the absence of stimuli, add 250-300 μl of fresh D10 media to chamber, replace cover and incubate in humidified 5% CO2 incubator at 37 °C. Do not let the cells in the chamber slide become dry at any point in the protocol as it will kill the parasites and host cells.
- If applicable, activate cells by dilution of stimulating agent or inhibitors in fresh D10 medium (250-300 μl/chamber).
- To activate macrophages, add LPS (10 ng/ml) and/or recombinant murine IFN-γ (100 U/ml).
- To inhibit iNOS while activating macrophages, add LPS (10 ng/ml), recombinant murine IFN-γ (100 U/ml), and aminoguanidine (1 mM).
- To challenge with the NO-donor sodium nitroprusside (SNP) add 100 μM of SNP.
- Macrophages can also be pre-activated up to 24 h prior to parasite invasion versus after parasite invasion although the effect on parasite replication is likely to be different as macrophages activated after parasite invasion are generally more permissive for parasite growth.
- To activate macrophages, add LPS (10 ng/ml) and/or recombinant murine IFN-γ (100 U/ml).
- Incubate for 24 h in a humidified incubator 37 °C and 5% CO2. If rate of parasite proliferation or doubling time needs to be determined kinetics can be performed instead of a single end point ie 6, 12, 24 and 36 h post parasite challenge.
- Following desired incubation; pour off media and fix macrophage monolayers with 250 μl per chamber with 4% paraformaldehyde (diluted to 4% with 1x PBS) for 20 min at 4 °C.
- Rinse chambers twice with 1x DPBS. Remove DPBS. Do not let chambers dry out during staining.
- Add 100 μl of blocking buffer with Triton X-100 detergent (see recipe) for 30 min to block and permeabilize the host cells and parasites so that they can be reached by antibodies.
- While waiting, dilute the unlabelled primary antibodies to the appropriate concentrations in blocking buffer:
- Rabbit polyclonal antibody against Toxoplasma gondii - 1:1,000 dilution. However, concentration of antibody needs to be optimized by the user. Other antibodies against Toxoplasma gondii or directly-conjugated fluorescence antibodies can be substituted.
- Pour off the blocking solution and add 100 μl of the primary antibody dilution to each well. Let stand for 1 h.
- While waiting, dilute the fluorescently labeled secondary antibodies to the appropriate concentrations in blocking buffer:
- Alexa Fluor® 488 goat anti-rabbit IgG (H + L) - 1:250 dilution. Optimal concentration of antibody may need to be optimized by user.
- Pour off the primary antibody and rinse each well three times with wash buffer (see Recipes) for 2 min per wash. Washes are performed by dumping out solution in slide and adding wash buffer 250-300 μl per chamber and letting solution sit for approximately 2 min.
- Add 100 μl of the secondary antibody dilution to each well and let stand for 1 h.
- Pour off the secondary antibodies and rinse each well four times with wash buffer for 2 min per wash.
- Wash each well two times with 1x DPBS for 2 min per wash.
- Mount the slides with Vectashield containing DAPI or other mounting media.
- Observe and analyze parasites in macrophages using a fluorescent microscope with a 100x phase or differential interference contrast (DIC) objective. The internal ultrastructure of the parasite is better with phase microscopy but DIC provides a more 3-dimensional perspective. We use a Zeiss inverted Axiovert 200 motorized microscope with a 100x objective, Zeiss filter sets 31, 34, 38, and 50, and Axiovision 4.3 software.
- To examine the replicative rate of the parasites, a time course analysis can be performed. To examine replication the number of parasites per each PV should counted. To examine whether parasites are killed by activation the number of parasites per macrophages at 1 h compared to later time points should be examined as well. If the number of parasites per macrophages is significantly reduced over time parasite cell death has occurred. Slides can also be co-stained with lysosomal associated membrane protein-1 using a species of antibody and flourochrome different from that used to detect parasites to evaluate the extent of phagosome-lysosome fusion (Mordue and Sibley, 1997).
- Examine at least 100 parasite-containing vacuoles per experiment and count the number of parasites per vacuole. We suggest counting two groups of 50 vacuoles or 100 vacuoles to obtain an average plus or minus standard deviation. Note the morphology of the parasites as some activation stimuli may result in parasite death and degradation or amorphous parasite-containing vacuoles.
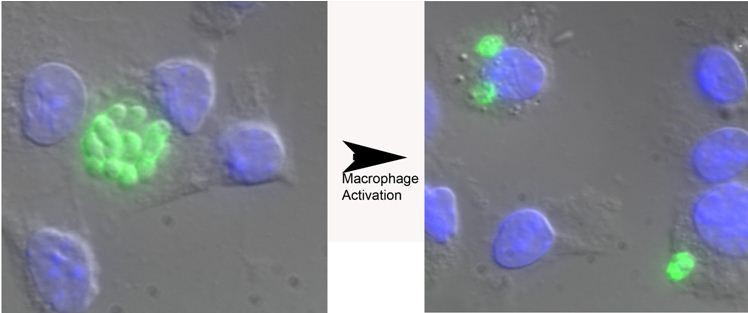
Figure 1. Toxoplasma gondii replication in naive versus murine bone marrow-derived macrophages activated with IFN-γ and LPS. Parasites are shown in green and the macrophage nucleus is in blue due to the DAPI in the Vectashield mounting media. The parasites in the vacuole in naive macrophages have greater than 8 parasites/vacuole while the parasites following activation of infected macrophages have only two parasites per vacuole. Other representative pictures are shown in References 1-3.
Recipes
- DPBS (we purchase it but the recipe is below)
2.7 mM KCl
1.5 mM KH2PO4
136.9 mM NaCl
8.9 mM Na2HPO4 (7H2O) - D10 media
500 ml DMEM
50 ml heat inactivated FCS (10% final concentration)
5 ml L-glutamine (final concentration 2 mM)
5 ml penicillin (100 U/ml final concentration) -streptomycin (100 μg/ml final concentration) - BMC media
D10 Media (recipe above)
20% L-929 cell conditioned medium - Blocking buffer
45 ml 1x DPBS
5 ml 10% fetal calf serum
100 μl Triton X100 (0.2% Triton X-100) - Wash buffer
49.5 ml 1x DPBS
0.5 ml 10% fetal calf serum (1% FCS final)
Protocol for L929 conditioned media
- Plate 5 x 105 L929 cells in a 150-cm2 tissue culture flask containing 50 ml of D10 medium.
- Grow cells to confluency in a humidified incubator with 5% CO2 at 37 °C and collect supernatant as L929 cells are beginning to round up and release from the flask. M-CSF is produced by L929 cells just prior to cell death).
- Collect supernatant and centrifuge it at 1,500 rpm (453 x g) (for 10 min to pellet any L929 cells remaining in the supernatant.
- Freeze in 40 ml aliquots in a 50 ml centrifuge tube prior to use (supernatant expands in freezer so centrifuge tube should not be too full prior to freezing).
Protocol for bone marrow isolation and bone marrow macrophage differentiation
- Sacrifice mouse by CO2 inhalation.
- Sterilize the abdomen and hind legs with 70% ethanol.
- Make an incision in the midline of the abdomen being careful not to penetrate the peritoneum. Pull back the skin above the peritoneum to fully expose the hind legs (both femur and tibia).
- Using sterile small dissecting scissors and forceps clean the femur and tibia of all muscle tissue. Separate the femur and tibia from the mouse by cutting at the hip and knee joint and knee and ankle joint and place in sterile petri dish containing 10 ml of sterile DPBS. Bones can be further cleared of muscle tissue at this point if needed.
- Cut at the end of both sides of the femur and tibia just enough to expose the bone marrow within.
- Fill a 10 ml syringe with a 25-gauge needle with 10 ml of DPBS. Insert needle into one end of the femur or tibia while holding bone firmly with forceps and flush bone marrow into a 50 ml centrifuge tube.
- Pipet the bone marrow cells up and down to disperse into a single cell suspension.
- Count the cells with a hemocytometer and adjust the concentration to 2 x 106 cells/ml in BMC media.
- Plate 10 ml of cells in each sterile bacteriological petri dish. The brand of bacteriological petri dish is important as macrophages adhere too tightly for easy removal on some brands and on tissue culture treated plastic.
- Incubate cells in a humidified incubator with 5% CO2 at 37 °C for one week. On day 5 add an additional 10 ml of BMC media to the media already in the petri dish. This is to provide fresh M-CSF but to leave in any macrophage-derived growth factor already produced by the macrophages in the petri dish.
- For macrophage removal remove supernatant and rinse the macrophages in the petri dish twice with DPBS without calcium and magnesium. Add 5 ml of DPBS without calcium and magnesium and place cells at 4 °C or in a refrigerator for 15-20 min. Use a sterile transfer pipette or serological pipette to dislodge macrophages off the petri plate (hold plate at slant and flush bottom of the plate. Successful dislodgement of macrophages will be visible as obvious clearance of cells from bottom of plate.
- Pipette macrophage-containing supernatant in 50 ml centrifuge tube and centrifuge at 1,500 rpm (453 rcf) for 10 min. Pour off supernatant and resuspend pellet in 10 ml of D10 media. Count cells using a hemocytometer and adjust to a concentration of 3 x 105 cells per ml. Add 250-300 μl to each chamber of an 8 well chamber slide. Replace cover that comes with chamber slide and Incubate cells in a humidified incubator with 5% CO2 at 37 °C overnight to let cells adhere (two hours is sufficient if cells need to be used the same day).
Acknowledgments
This work was adapted from a protocol we used for studies published in Skariah et al. (2012). The studies were funded by a grant to D.G. Mordue from the National Institute of Health (NIH) 1R01 AI 072028.
References
- Mordue, D. G. and Sibley, L. D. (1997). Intracellular fate of vacuoles containing Toxoplasma gondii is determined at the time of formation and depends on the mechanism of entry. J Immunol 159(9): 4452-4459.
- Pollard, A. M., Skariah, S., Mordue, D. G. and Knoll, L. J. (2009). A transmembrane domain-containing surface protein from Toxoplasma gondii augments replication in activated immune cells and establishment of a chronic infection. Infect Immun 77(9): 3731-3739.
- Skariah, S., Bednarczyk, R. B., McIntyre, M. K., Taylor, G. A. and Mordue, D. G. (2012). Discovery of a novel Toxoplasma gondii conoid-associated protein important for parasite resistance to reactive nitrogen intermediates. J Immunol 188(7): 3404-3415.
Article Information
Copyright
© 2012 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Iaconetti, E., Lynch, B., Kim, N. and Mordue, D. G. (2012). Determination of Toxoplasma gondii Replication in Naïve and Activated Macrophages. Bio-protocol 2(22): e289. DOI: 10.21769/BioProtoc.289.
Category
Immunology > Host defense > Murine
Immunology > Immune cell isolation > Macrophage
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.