- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Generation of Caenorhabditis elegans Transgenic Animals by DNA Microinjection


Published: Vol 7, Iss 19, Oct 5, 2017 DOI: 10.21769/BioProtoc.2565 Views: 16002
Reviewed by: Jyotiska ChaudhuriManish ChamoliAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2009
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Microinjection is the most frequently used tool for genetic transformation of the nematode Caenorhabditis elegans, facilitating the transgenic expression of genes, genome editing by the clustered regularly interspersed short palindromic repeats (CRISPR)-Cas9 system, or transcription of dsRNA for RNA intereference (RNAi). Exogenous DNA is delivered into the developing oocytes in the germline of adult hermaphrodites, which then generate transgenic animals among their offspring. In this protocol, we describe the microinjection procedure and the subsequent selection of transgenic progeny.
Keywords: Caenorhabditis elegansBackground
In C. elegans, DNA transformation by microinjection is commonly used to produce transgenic animals that over-express or ectopically express genes, which can be fused to tags (e.g., green fluorescent protein [GFP]), allowing for the phenotypic rescue of mutants, and/or the analysis of localization and function of proteins (Carter et al., 1990; Chalfie et al., 1994; Mello and Fire, 1995). The advent of the clustered regularly interspersed short palindromic repeats (CRISPR)-Cas9 system requires microinjection to achieve highly specific genome editing by introducing point mutations or insertion/deletion mutations (summarized in Dickinson and Goldstein, 2016). Further, the technique is applied for the inducible and/or tissue-specific in vivo transcription of dsRNA to facilitate heritable RNA interference (RNAi) (Tavernarakis et al., 2000). The genetic material is injected into the distal gonad syncytium of adult hermaphrodite animals where it enters the developing oocytes (Figure 1). The exogenous DNA is arranged into large extrachromosomal arrays consisting of 50 to 300 copies, which are inherited in a non-Mendelian fashion to the following generations. Next-generation progeny (F1) carry the extrachromosomal array with a varying probability (between 5 to 80% [Stinchcomb et al., 1985; Mello et al., 1991]). Transgenic animals are identified amongst the offspring by the use of co-transformation markers, which are injected along with the DNA of interest (Table 1). Most commonly used are the pharyngeal expression of green fluorescent protein (GFP) or red fluorescence (mCherry), or the dominant rol-6(su1006) allele, which induces a distinct rolling phenotype in transgenic F1 progeny (Mello et al., 1991; Tabara et al., 1996; Frokjaer-Jensen et al., 2008).
Table 1. Commonly used co-transformation markers with distinct phenotypes for selection of transgenic animals upon microinjection in C. elegans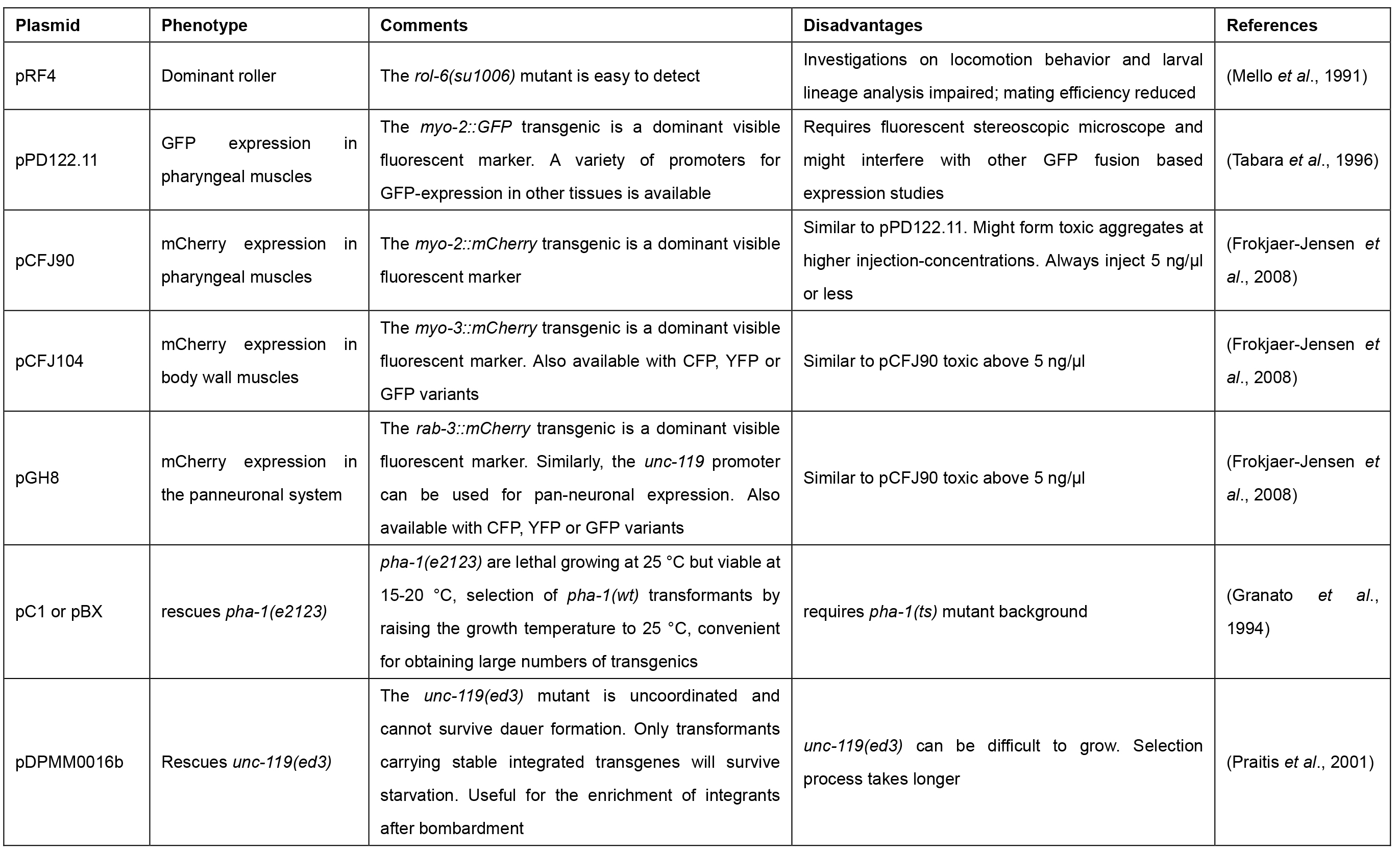
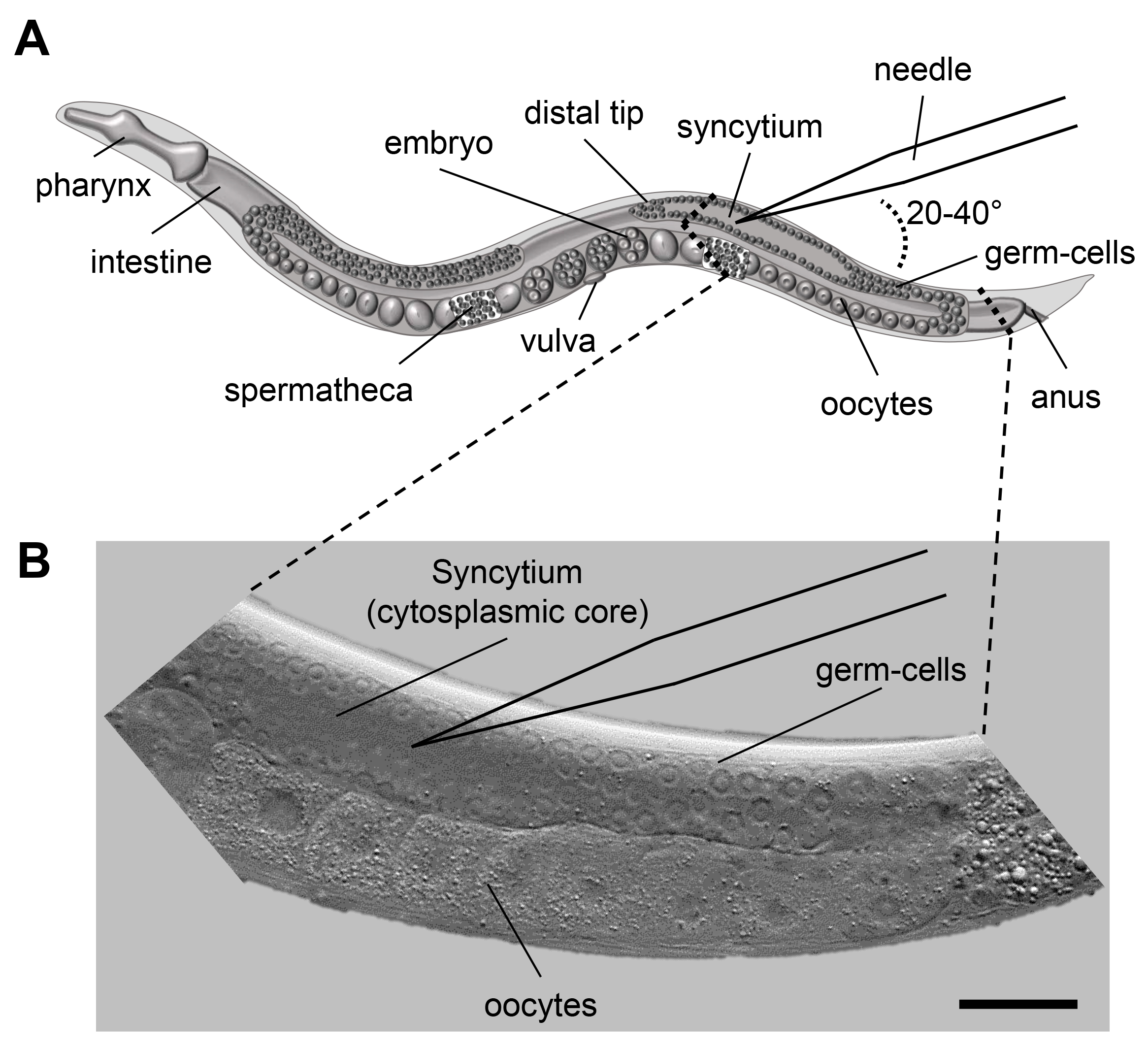
Figure 1. Microinjection scheme for C. elegans. A. Scheme of an adult C. elegans displaying the major organs including the pharynx, intestine and the gonad. When microinjecting C. elegans the injection capillary needs to be inserted in the syncytium (cytoplasmic core) of the distal gonad. The inlay indicates the area of interest for injection. B. DIC image of the area indicated in 1A. The nuclei of the germ-cells are clearly visible and surround the syncytium. Size bar corresponds to 50 µm.
Materials and Reagents
- Sterile pipette tips
- Microloader pipette tips (Eppendorf, catalog number: 5242956003 )
- Microinjection capillaries (Eppendorf Femtotips II, 0.5 µm inner and 0.7 µm outer diameter) (Eppendorf, catalog number: 930000043 )
- Microscope slides, 76 x 26 mm (Carl Roth, catalog number: 0656.1 )
- Microscope cover glasses, 24 x 60 mm (VWR, catalog number: 631-1575 )
- Tape (~1 mm thickness)
- Greiner Petri dishes (60 x 15 mm) (Sigma-Aldrich, catalog number: P5237 )
- Glass Pasteur Pipettes, disposable, 150 mm (BRAND, catalog number: 747715 )
- C. elegans animals (e.g., available from Caenorhabditis Genetics Center [CGC], University of Minnesota, MN, USA)
- Escherichia coli OP50 strain (obtained from the Caenorhabditis Genetics Center)
- DNA of interest for injection (plasmid, fosmid, PCR fragment, or similar) and plasmid containing a co-transformation marker (see Table 1)
- Miniprep Kit for DNA purification (e.g., QIAGEN-tip 20) (QIAGEN, catalog number: 10023 )
- Halocarbon oil 700 (Sigma-Aldrich, catalog number: H8898-100ML )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S9888 )
- Peptone (BD, BactoTM, catalog number: 211677 )
- Streptomycin sulfate salt (Sigma-Aldrich, catalog number: S6501 )
- Agar (Sigma-Aldrich, catalog number: 05040 )
- 100% ethanol
- Calcium chloride dehydrate (CaCl2·2H2O) (Sigma-Aldrich, catalog number: C5080 )
- Magnesium sulfate (MgSO4) (Sigma-Aldrich, catalog number: M7506 )
- Cholesterol stock solution (SERVA Electrophoresis, catalog number: 17101.01 )
- Nystatin stock solution (Sigma-Aldrich, catalog number: N3503 )
- Potassium dihydrogen phosphate (KH2PO4) (Carl Roth, catalog number: P018.1 )
- Di-Sodium hydrogen phosphate (Na2HPO4) (Carl Roth, catalog number: T876.1 )
- Di-Potassium hydrogen phosphate (K2HPO4) (Carl Roth, catalog number: 5066.1 )
- Agarose (Biozym Scientific, catalog number: 840004 )
- OP50 seeded NGM agar plates (see Recipes)
- 2% agarose pads (see Recipes)
- Hairpin pick (see Recipes)
- M9 buffer (see Recipes)
Equipment
- Bunsen burner
- Cylindrical glass beaker
- Wormpick with platinum wire and pre-flattened tip (Genesee Scientific, catalog numbers: 59-AWP and 59-30P6 )
- Microwave
- Heat plate (e.g., IKA, model: C-MAG HP 4 , catalog number: 0003581600)
- Puller (optional; e.g., Sutter Instrument, model: P-97 )
- Autoclave
- Stereomicroscope (e.g., Leica Microsystems, model: Leica M80 ), optionally fluorescent stereomicroscope (e.g., Leica Microsystems, model: Leica M165 FC)
- Microscope for Microinjection (ZEISS, model: Axio Observer.A1 equipped with differential interference contrast [DIC] prisms, gliding table, objective 10x/0.3 M27, objective 40x/0.75 M27, optionally camera AxioCam ERc 5 sec and monitor for observation and demonstration purposes)
- Micromanipulator (e.g., Eppendorf InjectMan 4) (Eppendorf, catalog number: 5192000019 )
- Microinjector unit (e.g., Eppendorf FemtoJet 4i) (Eppendorf, catalog number: 5252000013 )
- Microcentrifuge (e.g., Eppendorf Centrifuge 5424) (Eppendorf, model: 5424 , catalog number: 5404000014)
- Spectrophotometer (e.g., Thermo Fisher Scientific, model: NanoDropTM 8000 , catalog number: ND-8000-GL)
Procedure
- Preparing the DNA and the injection unit
- Follow the instructions of the QIAGEN-tip 20 miniprep kit for DNA purification (see Notes).
- At the final step, elute the DNA in distilled H2O and determine the concentration with a spectrophotometer.
- Prepare a mix of 50 ng/µl of the DNA of interest and the co-transformation marker each (see Table 1) in a 100 µl volume.
- Centrifuge the solution in a microcentrifuge at maximum speed for 15 min, preferably at 4 °C. This step is necessary to precipitate floating particles in the DNA solution to prevent clogging of the microinjection capillary.
- Pipette 5 µl from top of the DNA solution to a new reaction tube and directly use it for microinjection. Any left-over can be frozen at -20 °C for later use.
- A few minutes prior to the injection procedure, switch on the microscope for microinjection, the microinjector unit and the micromanipulator (overview in Figure 2). Follow the instructions of the microinjector unit to pressurize and adjust the system. Set the ‘inject’ function to release a volume of approx. 10 picoliter (pl).

Figure 2. Basic components of a setup for C. elegans microinjection. A. Overview over the setup and all its components: (a) light microscope with a 10x and a 40x objective and DIC optics, (b) microinjector unit for pressure appliance, (c1) micromanipulator control for all three dimensions (x, y, z axis) with fine and coarse control, (c2) micromanipulator-controlled capillary (needle) holder, (d) gliding table to flexibly move immobilized animals into the needle, (e) stereomicroscope for mounting the animals on agarose pads and recovery after injection, (f) optionally a monitor, connected to a camera for recording and demonstration purposes, (g) glass capillary (injection needle). B. Detailed view on the gliding table (d), the needle holder (c2) and the needle (g) attached. C. Close-up on the injection needle (Femtotips II, Eppendorf), which can be filled by microloader pipette tips. - Use the microloader pipette tips to pipette 1 µl of the clean DNA solution into a microinjection capillary (see Notes). This volume is enough to inject more than a hundred animals.
- Carefully remove the cover of the capillary and screw it into the needle holder of the micromanipulator. The capillary force will collect the solution in the tip of the needle.
- Follow the instructions of the QIAGEN-tip 20 miniprep kit for DNA purification (see Notes).
- Preparing the animals for injection
- One day prior to the injection procedure, use a stereoscope and a wormpick to transfer 40 L4 stage animals from a mixed population to freshly OP50-seeded NGM agar plates (see Recipes).
- Grow the animals at 20 °C overnight into young gravid adults with a fully developed germline (Figure 1A).
- One hour prior to the injection procedure shift the animals to 15 °C. This reduces the agility of animals, making them easier to handle for the injection.
- Place a drop of about 50 µl halocarbon oil on a 2% agarose pad (see Recipes) next to the agarose area. Under the stereoscope, transfer 10-20 worms into the oil (dipping the worm pick into the oil makes it easier to transfer the worms). Let the animals move in the oil for 5-10 min to remove bacteria that are attached to their cuticle as bacterial remnants can quickly clog the microinjection capillary.
- Place another small drop of about 20 µl halocarbon oil on the surface of the agar pad.
- Use the hairpin pick (see Recipes) to transfer one animal from the halocarbon oil next to the agarose into the drop on the surface of the agarose pad.
- Use the maximum magnification of the stereoscope to gently press the animal to the agarose surface with the hairpin pick. The animals will attach to the dry agarose surface (Figure 3).
- Place the agarose pad on the gliding table of the microscope for microinjection and swiftly commence with the injection procedure, as the animals will desiccate within approx. 10-15 min.
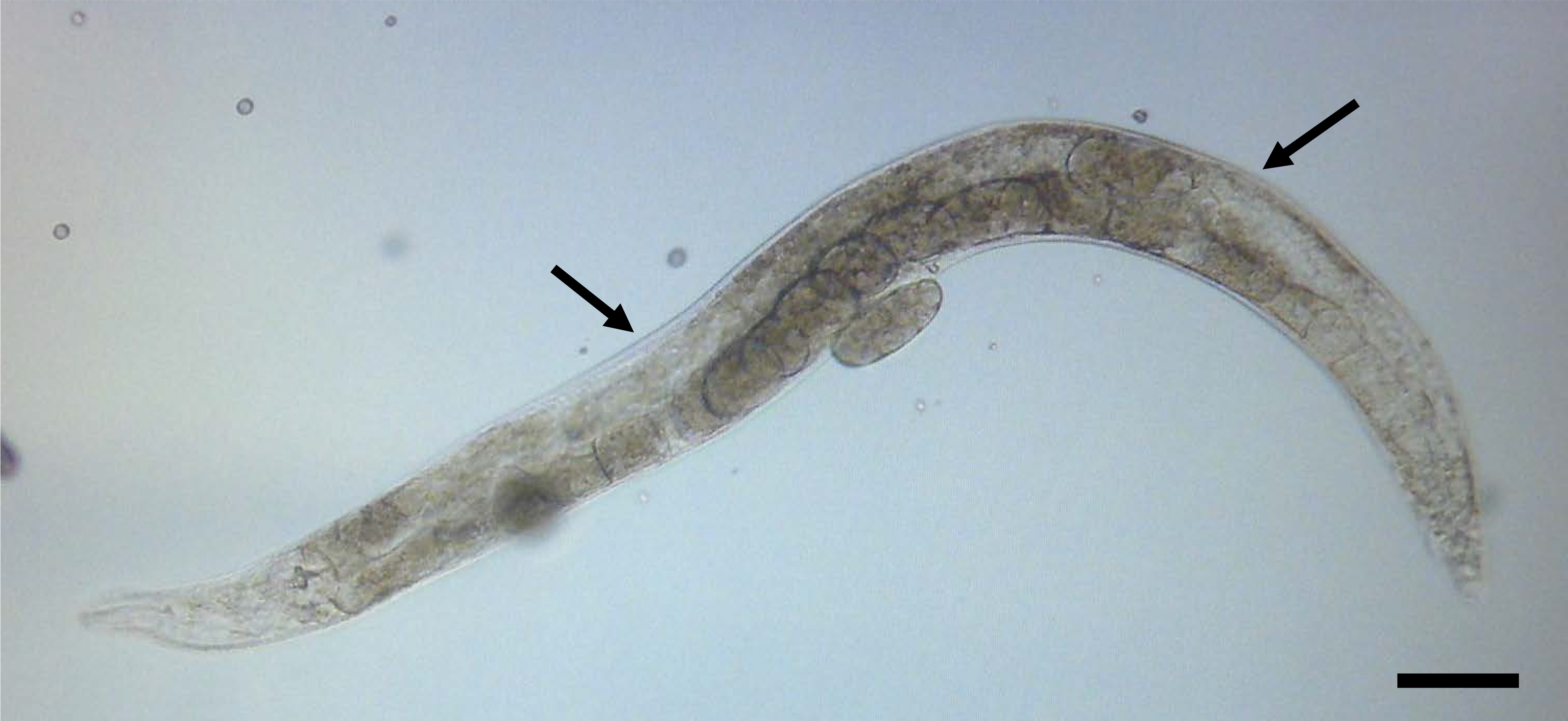
Figure 3. An animal immobilized on a 2% agarose pad and ready for injection. The arrows indicate the preferred sites for injection in the gonad. Scale bar is 50 µm.
- One day prior to the injection procedure, use a stereoscope and a wormpick to transfer 40 L4 stage animals from a mixed population to freshly OP50-seeded NGM agar plates (see Recipes).
- Injection procedure
- Use the 10x objective to localize the immobilized animal in the halocarbon oil drop on the agarose pad.
- Use the micromanipulator in ‘coarse’ mode to move the tip of the injection capillary along the x-y axis in close proximity of the worm and then use the ‘fine’ mode of the micromanipulator z-axis control to roughly bring the tip of the needle in focus (Video 1).
- Adjust the position of the worm to a 20-40° angle relative to the needle by turning the gliding table (Figure 1A). The steep angle is necessary to prevent injuries in the cuticle.
- Switch to the 40x objective and focus on the cytoplasmic core of the distal germline of the animal. Then bring the tip of the needle close and in focus by using the micromanipulator in ‘fine’ mode.
- Move the animal by gently tapping the gliding table towards the tip of the needle. We move the animal into the needle, not the other way around, as this is easier and more precisely to handle. The needle tip has to be placed inside the cytoplasmic core of the distal gonad (Figures 1A and 1B).
- Press the ‘Inject’ button on the Microinjector unit to release a specified volume of DNA solution into the gonad. The liquid will slightly blow up the gonad (Video 1).
 Video 1. The injection procedure. The video shows (1) The localization of the needle and test release of injection mix, (2) Localization of the target animal and placing it into position for injection by rotating the gliding table, (3) Adjusting the needle position to target the gonad, (4) Switching to the 40x objective and centralize the position of the needle, (5) Focusing on the syncytium of the distal gonad, (6) Moving the animal into the needle and injecting the injection mix, as well as three more examples for the procedure.
Video 1. The injection procedure. The video shows (1) The localization of the needle and test release of injection mix, (2) Localization of the target animal and placing it into position for injection by rotating the gliding table, (3) Adjusting the needle position to target the gonad, (4) Switching to the 40x objective and centralize the position of the needle, (5) Focusing on the syncytium of the distal gonad, (6) Moving the animal into the needle and injecting the injection mix, as well as three more examples for the procedure. - Gently move the gliding table in the opposite direction to remove the needle from the animal.
- Use the micromanipulator to move the needle up along the z-axis but leave it in the same x-y position for the next rounds of injection. Remove the agarose pad from the gliding table.
- Under the stereoscope, pipette a drop of 10 µl M9 buffer (see Recipes) on the injected animal to remove it from the agarose pad. Let the animal recover for 2 min in the M9 buffer before transferring it with a hairpin pick to an OP50 seeded NGM agar plate.
- Repeat the injection procedure with a new animal (go to step B5). Inject 20-25 animals in total. With increasing experience and injection speed, several animals can be immobilized on the agarose pad and injected sequentially. In that case, align the animals close to each other under the stereoscope. Upon successfully injecting the first animal move the injection needle out of focus up along the z-axis, move the gliding table to focus on the next animal (switching to the 10x objective might be required) and repeat the injection procedure.
- Use the 10x objective to localize the immobilized animal in the halocarbon oil drop on the agarose pad.
- Selecting transgenic animals and maintaining stabile transgenic lines
- After the injection procedure grow the animals for 3-5 days at 20 °C. During that time, monitor the F1 progeny for the distinct phenotypic features caused by the expression of the co-transformation marker (see Table 1). This might require observation under a fluorescent stereomicroscope to detect expression of fluorescent proteins. The injection efficiency can broadly vary between 1-10% and more.
- Single out transgenic F1 progeny as soon as they reach the L4 or adult stage by picking them each to a new OP50 seeded NGM plates with a wormpick. Keep maintaining them at 20 °C and monitor the F2 generation for the transgenic phenotype. Approx. 5-10% of the transgenic F1 animals will stably inherit the extrachromosomal array in a non-Mendelian fashion. Hence, it is recommended to monitor at least 30-50 F1 transgenics for stabile inheritance.
- Stably inheriting transgenic lines have to be maintained by picking 10-30 transgenic animals to fresh OP50 seeded NGM plates every 3-5 days. The transmission efficiency across generations can vary as much as 5-80%. Maintain at least three independent stable lines for phenotypic analysis when over-expressing or ectopically expressing genes or dsRNA constructs. For CRISPR/Cas9 side-directed mutagenesis other maintenance and selection criteria will apply, which are summarized elsewhere (Dickinson and Goldstein, 2016).
- After the injection procedure grow the animals for 3-5 days at 20 °C. During that time, monitor the F1 progeny for the distinct phenotypic features caused by the expression of the co-transformation marker (see Table 1). This might require observation under a fluorescent stereomicroscope to detect expression of fluorescent proteins. The injection efficiency can broadly vary between 1-10% and more.
Notes
- DNA samples: C. elegans can be transformed with any type of DNA including plasmid, fosmid or cosmid DNA, phage DNA, genomic DNA or linear DNA obtained from a PCR reaction. Generally, any commercial purification protocol can be used to clean up the DNA. We do not recommend protocols such as boiling lysis or phenol-chloroform extraction as the resulting DNA solution might contain chemicals that are toxic for the animals. A critical factor for successful transformation is the concentration of the DNA: We suggest to use 50-100 ng/µl per injected construct, which is sufficient to form an extrachromosomal array. In some cases, an extrachromosomal array can lead to lethality, which is often based on the dose-dependency of expression. This issue can often be overcome by lowering the concentration of the DNA construct, which leads to the formation of a smaller extrachromosomal array containing less gene copies (Mello et al., 1991). Generally, we inject DNA dissolved in distilled H2O or 1x TE buffer. A special microinjection buffer has been suggested to be advantageous in some cases, e.g., for direct injection into maturing oocyte nuclei (Fire, 1986).
- Microinjection capillaries: The injection needles can also be prepared with a needle puller (e.g., P-97 , Sutter Instrument, Novato, CA, USA). The main advantage is that the diameter and length of the tip can be adjusted to the desired dimensions. However, the tip of the needle has to be opened by carefully breaking it on the surface of the agarose pad or by dipping it into hydrofluoric acid.
- Extrachromosomal arrays obtained from microinjection can be integrated in the genome by causing chromosomal breaks through mutagenic chemicals (Ethylmethane Sulphonate, EMS) or ionizing radiation (Rieckher et al., 2009).
- The microinjection protocol can easily be adapted to inject a number of other chemicals, e.g., proteins, drugs or small RNA molecules in the gonad or other distinct tissues, such as the intestine.
Recipes
- OP50 seeded NGM agar plates
- For 1 L NGM agar, combine 3 g NaCl, 2.5 g Bacto peptone, 0.2 g streptomycin and 17 g agar. Autoclave for 30 min including stir bar and cool to 55 °C while stirring
- Next to a Bunsen burner, add 1 ml 1 M CaCl2 (stock: 14.7 g CaCl2 in 100 ml distilled H2O, autoclaved), 1 ml 1 M MgSO4 (stock: 12.03 g CaCl2 in 100 ml distilled H2O, autoclaved), 1 ml cholesterol (5 mg/ml in 100% ethanol), 1 ml nystatin (10 mg/ml in 70% ethanol) and 1 ml KPO4 (stock: 102.2 g KH2PO4 and 57.06 g K2HPO4 in 1 L distilled H2O, autoclaved)
- Use 60 mm Petri dishes and pour 11.5 ml of NGM agar per plate (adjust volume to size of Petri dishes). Let cool and harden for some hours
- Next to the Bunsen burner, streak a drop (~200 µl) of an Escherichia coli OP50 liquid culture in the center of each plate and grow at 37 °C for 8 h or at room temperature overnight
- For 1 L NGM agar, combine 3 g NaCl, 2.5 g Bacto peptone, 0.2 g streptomycin and 17 g agar. Autoclave for 30 min including stir bar and cool to 55 °C while stirring
- 2% agarose pads for worm immobilization (prepare several in parallel)
- Weigh 2.0 g agarose in a cylindrical glass beaker
- Add 100 ml distilled H2O
- Heat in a microwave until close to boiling. Take out, stir with a pipette tip and boil again. Repeat until the agarose is dissolved
- On a clean bench, fix a microscope cover glass with two stripes of tape
- Put a drop (ca. 50 µl) of fresh 2% agarose solution in the middle of the cover glass
- Take a microscope slide and place it on top of the agarose drop. Gently press down to flatten the drop. The tape serves as spacer to give the agarose pad a specific thickness (Figure 4)
- Let the agarose harden for 30 sec. And remove the microscope slide
- Set a heat plate to 100 °C and put the agarose pad on top for approx. 1 min to dry the agarose. The slides can be prepared in advance and stored indefinitely

Figure 4. Preparing of 2% agarose pad for microinjection. A. Use two stripes of tape to fix a coverglass to a clean work bench; B. Place a drop of about 50 µl freshly prepared 2% agarose in distilled H2O in the middle of the coverglass; C. Put a microscope slide on top of the agarose drop and gently press down to flatten it. Let the agarose solidify and remove the microscope slide.
- Weigh 2.0 g agarose in a cylindrical glass beaker
- Hairpin pick
Remove a hair from your eyebrows and glue it to the tip of a toothpick or a glass Pasteur pipette (Figure 5). Best is to prepare many of them with hair of different thickness and test the handling convenience. While using, sterilize the hairpin by dipping it into a 70% ethanol solution
Figure 5. Preparing a hairpin pick for gentle manipulation of C. elegans. Remove a hair from your eyebrow and glue it to the tip of a glass Pasteur pipette or a toothpick. - M9 buffer
22 mM KH2PO4
22 mM Na2HPO4
85 mM NaCl
1 mM MgSO4
Combine in distilled H2O
Sterilize by autoclaving for 15 min at 121 °C
Acknowledgments
This work was funded by grants from the European Research Council (ERC), the European Commission 7th Framework Programme. This work has been adapted from Rieckher M., N. Kourtis N., Pasparaki A. and Tavernarakis N. (2009). ‘Transgenesis in Caenorhabditis elegans.’ Methods Mol Biol 561: 21-39.
References
- Carter, P. W., Roos, J. M. and Kemphues, K. J. (1990). Molecular analysis of zyg-11, a maternal-effect gene required for early embryogenesis of Caenorhabditis elegans. Mol Gen Genet 221(1): 72-80.
- Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W. and Prasher, D. C. (1994). Green fluorescent protein as a marker for gene expression. Science 263(5148): 802-805.
- Dickinson, D. J. and Goldstein, B. (2016). CRISPR-based methods for Caenorhabditis elegans genome engineering. Genetics 202(3): 885-901.
- Fire, A. (1986). Integrative transformation of Caenorhabditis elegans. EMBO J 5(10): 2673-2680.
- Frokjaer-Jensen, C., Davis, M. W., Hopkins, C. E., Newman, B. J., Thummel, J. M., Olesen, S. P., Grunnet, M. and Jorgensen, E. M. (2008). Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet 40(11): 1375-1383.
- Granato, M., Schnabel, H. and Schnabel, R. (1994). pha-1, a selectable marker for gene transfer in C. elegans. Nucleic Acids Res 22(9): 1762-1763.
- Mello, C. and Fire, A. (1995). DNA transformation. Methods Cell Biol 48: 451-482.
- Mello, C. C., Kramer, J. M., Stinchcomb, D. and Ambros, V. (1991). Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J 10(12): 3959-3970.
- Praitis, V., Casey, E., Collar, D. and Austin, J. (2001). Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics 157(3): 1217-1226.
- Rieckher, M., Kourtis, N., Pasparaki, A. and Tavernarakis, N. (2009). Transgenesis in Caenorhabditis elegans. Methods Mol Biol 561: 21-39.
- Stinchcomb, D. T., Shaw, J. E., Carr, S. H. and Hirsh, D. (1985). Extrachromosomal DNA transformation of Caenorhabditis elegans. Mol Cell Biol 5(12): 3484-3496.
- Tabara, H., Motohashi, T. and Kohara, Y. (1996). A multi-well version of in situ hybridization on whole mount embryos of Caenorhabditis elegans. Nucleic Acids Res 24(11): 2119-2124.
- Tavernarakis, N., Wang, S. L., Dorovkov, M., Ryazanov, A. and Driscoll, M. (2000). Heritable and inducible genetic interference by double-stranded RNA encoded by transgenes. Nat Genet 24(2): 180-183.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Rieckher, M. and Tavernarakis, N. (2017). Generation of Caenorhabditis elegans Transgenic Animals by DNA Microinjection. Bio-protocol 7(19): e2565. DOI: 10.21769/BioProtoc.2565.
Category
Molecular Biology > DNA > Transformation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.