- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Peroxisome Motility Measurement and Quantification Assay



Published: Vol 7, Iss 17, Sep 5, 2017 DOI: 10.21769/BioProtoc.2536 Views: 8293
Reviewed by: Pengpeng LiThirupugal GovindarajanAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2017

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Organelle movement, distribution and interaction contribute to the organisation of the eukaryotic cell. Peroxisomes are multifunctional organelles which contribute to cellular lipid metabolism and ROS homeostasis. They distribute uniformly in mammalian cells and move along microtubules via kinesin and dynein motors. Their metabolic cooperation with mitochondria and the endoplasmic reticulum (ER) is essential for the β-oxidation of fatty acids and the synthesis of myelin lipids and polyunsaturated fatty acids. A key assay to assess peroxisome motility in mammalian cells is the expression of a fluorescent fusion protein with a peroxisomal targeting signal (e.g., GFP-PTS1), which targets the peroxisomal matrix and allows live-cell imaging of peroxisomes. Here, we first present a protocol for the transfection of cultured mammalian cells with the peroxisomal marker EGFP-SKL to observe peroxisomes in living cells. This approach has revealed different motile behaviour of peroxisomes and novel insight into peroxisomal membrane dynamics (Rapp et al., 1996; Wiemer et al., 1997; Schrader et al., 2000). We then present a protocol which combines the live-cell approach with peroxisome motility measurements and quantification of peroxisome dynamics in mammalian cells. More recently, we used this approach to demonstrate that peroxisome motility and displacement is increased when a molecular tether, which associates peroxisomes with the ER, is lost (Costello et al., 2017b). Silencing of the peroxisomal acyl-CoA binding domain protein ACBD5, which interacts with ER-localised VAPB, increased peroxisome movement in skin fibroblasts, indicating that membrane contact sites can modulate organelle distribution and motility. The protocols described can be adapted to other cell types and organelles to measure and quantify organelle movement under different experimental conditions.
Keywords: Peroxisome motilityBackground
An important feature of eukaryotic cells is the presence of membrane-bound compartments (organelles), which create distinct optimised environments to promote various metabolic reactions required to sustain life. For the entire cell to function as a unit, coordination and cooperation between specialized organelles must take place. This requires a dynamic spatial organization, which allows the movement of organelles to areas of greatest metabolic need, the positioning of organelles in those areas, and the interaction with other compartments, which permits metabolic cooperation and communication amongst organelles. This is often mediated through interorganellar membrane contacts, whereby two organelles come into close apposition (Prinz, 2014; Eisenberg-Bord et al., 2016).
Peroxisomes are multifunctional organelles that play pivotal cooperative roles in the metabolism of cellular lipids and reactive oxygen species (ROS) and are thus essential for human health and development (Islinger and Schrader, 2011; Wanders et al., 2015; Waterham et al., 2016). Peroxisomes interact with many organelles involved in cellular lipid metabolism such as the endoplasmic reticulum (ER), mitochondria, lipid droplets or lysosomes (Schrader et al., 2013 and 2015). We revealed that the peroxisomal tail-anchored membrane proteins ACBD5 and ACBD4 directly interact with tail-anchored VAPB at the ER (Costello et al., 2017a; 2017b and 2017c). This interaction links both organelles together and allows transfer of lipids between them (Costello et al., 2017b; Hua et al., 2017). Whereas in plants and yeast, peroxisomes move along the actin cytoskeleton by interacting with myosin motors (Jedd and Chua, 2002; Fagarasanu et al., 2009), in mammalian cells (and filamentous fungi), peroxisomes use microtubules and dynein/kinesin motors to distribute uniformly within the cell and to reach new neighbourhoods (Schrader et al., 1996 and 2003; Guimaraes et al., 2015; Lin et al., 2016). We also found that ACBD5-VAPB interaction, which tethers peroxisomes to the ER, influences peroxisome motility (Costello et al., 2017b). Using the peroxisome motility measurement and quantification assay, we showed that loss of ACBD5, which resulted in reduced peroxisome-ER association, also increased peroxisome movement (Costello et al., 2017b). Our data indicate that organelle contact sites can modulate peroxisome (organelle) distribution and motility.
Peroxisomes are highly versatile organelles, which respond to environmental stimuli with changes in their number, size, and enzyme composition (Islinger et al., 2010). Certain stress conditions, in particular in plants, can lead to changes in the motile behaviour of peroxisomes and altered distribution (Rodríguez-Serrano et al., 2009). There is currently great interest in the measurement of peroxisome (organelle) motility and its quantification in order to understand the fundamental principles of organelle distribution, its regulation and role in organelle interaction and metabolic cooperation. Understanding these mechanisms is not just important for comprehending fundamental physiological processes but also for understanding pathogenic processes in disease etiology (Ferdinandusse et al., 2017; Yagita et al., 2017).
Materials and Reagents
- 10 cm culture dishes (Greiner Bio One International, catalog number: 664160 )
- Microporation tips
- 15 ml centrifuge tubes (Greiner Bio One International, catalog number: 188271 )
- 1.5 ml microcentrifuge tubes (Greiner Bio One International, catalog number: 616201 )
- 3.5 cm round glass bottom dishes (Cellview, Greiner Bio One International, catalog number: 627861 )
- Serological pipettes, 10 ml (Greiner Bio One International, catalog number: 607180 )
- Mammalian cell line of interest, here: human skin fibroblasts (see Note 1)
- Peroxisome marker (fluorescent reporter with a C-terminal peroxisomal targeting signal, e.g., EGFP-SKL plasmid [Schrader et al., 2000])
- Optional: siRNA for silencing of candidate genes/proteins in mammalian cells, here: ACBD5 (Ambion, catalog number: s40666 ); control scrambled siRNA (GE Healthcare Dharmacon, catalog number: D-001206-14-05 ) (50-100 µM stock) (in sterile, RNase/DNase-free water or buffer supplied by manufacturer)
- 70% (v/v) ethanol
- TrypLETM Express solution (1x) (Thermo Fisher Scientific, GibcoTM, catalog number: 12604013 ) (store at 4 °C)
- Immersion oil (Olympus, catalog number: IMMOIL-F30CC )
- Dulbecco’s modified Eagle medium (DMEM) high glucose (4.5 g/L) (Thermo Fisher Scientific, GibcoTM, catalog number: 11965092 ) for complete growth medium for cell culture of human skin fibroblasts
- Fetal bovine serum (FBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 10082147 )
- Penicillin/streptomycin solution (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 )
- Sodium chloride (NaCl)
- Potassium chloride (KCl)
- Sodium phosphate dibasic (Na2HPO4)
- Potassium phosphate dibasic (K2HPO4)
- Complete growth medium for cell culture of human skin fibroblasts (see Recipes)
- Phosphate-buffered saline (1x PBS) (see Recipes)
Equipment
- Pipetting aid (Greiner Bio One International, model: Sapphire MAXIPETTE, catalog number: 847070 )
- Class II biological safety cabinet/tissue culture hood (Faster, model: SafeFAST Top 209-D, catalog number: F00000050000 ) (see Note 2)
- Humidified CO2 incubator (95% air, 5% CO2, 37 °C) (Thermo Fisher Scientific, Thermo ScientificTM, model: HeracellTM 240i , catalog number: 510263330)
- Inverted light microscope (phase contrast) (ZEISS, model: Primo VertTM )
- 37 °C water bath (Grant Instruments, model: JBA12 )
- TC20TM Automated Cell Counter (Bio-Rad Laboratories, catalog number: 145-0101 )
- Vacuum aspiration system (Fisher Scientific, catalog number: 11636620)
Manufacturer: INTEGRA Biosciences, catalog number: 158320 . - Table top centrifuge equipped with a swing-out rotor for 15-ml conical tubes (Thermo Fisher Scientific, Thermo ScientificTM, model: HeraeusTM BiofugeTM StratosTM , catalog number: 75005282)
- Microcentrifuge (Thermo Fisher Scientific, Thermo ScientificTM, model: HeraeusTM PicoTM 17 , catalog number: 75002491)
- Microporator Neon® Transfection System (Thermo Fisher Scientific, InvitrogenTM, catalog number: MPK5000S )
- Spinning disk microscope with controlled temperature-CO2 chamber and objective warmer
Note: An Olympus IX81 microscope (Olympus, model: IX81 ) equipped with a Yokogawa CSUX1 spinning disk (Yokogawa Electric, model: CSU-X1 ) head, CoolSNAP HQ2 CCD camera (Photometrics, model: CoolSNAP HQ2 ) and a UPlanSApo 60x/1.35 oil objective was used. Image acquisition was performed using a 488 nm solid state laser at 20% of max. intensity.
Software
- VisiView software (Visitron Systems, Germany)
- Fiji (ImageJ)
- Custom Python data analysis pipeline
Procedure
- Cell culture and transfection
- Perform all cell culture based work in a class II biological safety cabinet/tissue culture hood and disinfect the work surface and materials (e.g., pipettes) with 70% (v/v) ethanol.
- Grow mammalian cells of choice (here, human skin fibroblasts) in complete growth medium (see Recipes) (10 cm Ø cell culture dishes or cell culture flasks) in a humidified CO2 incubator (95% air, 5% CO2, 37 °C).
- For maintenance of cells, refresh the cell culture medium every 2-3 days and split the cells before they reach 100% confluency using standard cell culture procedures (see Note 3).
- One or two days prior to transfection split the cells so that they reach 70-80% confluency at the day of transfection (10 cm Ø cell culture dishes or cell culture flasks).
- Prior to transfection prepare 3.5 cm glass bottom dishes with complete growth medium (without antibiotics), and pre-incubate in the CO2 incubator at 37 °C.
- In addition, place the Neon® Microporation device in the biological safety cabinet. Fill a Neon tube with 3 ml of electrolyte buffer E and insert the tube into the pipette station. Set the transfection parameters (here, 1,700 V, 20 msec pulse width, 1 pulse) on the device (see Notes 4 and 5).
- For transfection, wash the cells (from 10 cm Ø cell culture dishes or cell culture flasks) once with 1x PBS (see Recipes) and incubate for 2-5 min with 1 ml of TrypLE Express at 37 °C.
- Resuspend detached cells in 9 ml of complete growth medium (without antibiotics), and determine the cell number using a TC20TM Automated Cell Counter (see Note 6).
- For each transfection reaction use 1-2 x 105 cells (number of cells used with a 10 µl microporation tip) (see Note 7). Transfer the total number of cells for all transfections to a 15 ml tube and centrifuge for 3 min at RT in a table top centrifuge (500 x g).
- Resuspend the cell pellet in 1 ml of 1x PBS and transfer cells to a 1.5 ml microcentrifuge tube.
- Centrifuge cells in a microcentrifuge for 3 min at 500 x g and resuspend the pellet in buffer R. The amount of buffer R will depend on the number of transfections and the microporation tip used (e.g., 6 transfections with a 10 µl microporation tip–6 x 10 µl = 60 µl).
- For each transfection, pre-mix 1-2 µg of plasmid DNA (here, EGFP-SKL) and 50-100 nM of siRNA (optional) in a microcentrifuge tube and add 10 µl of cells resuspended in buffer R. To facilitate pipetting, mix enough reagents for at least 2 transfections in each microcentrifuge tube (see Note 8).
- Mount a 10 µl Neon tip onto the Neon pipette.
- Immerse the tip into the cell-DNA-siRNA mixture and slowly aspirate 10 µl of the sample. Avoid generating air bubbles in the tip.
- Insert the pipette into the E buffer–containing tube in the pipette station, and press start on the touch screen.
- After delivery of the electric pulse, quickly remove the pipette from the pipette station and immediately transfer the cells from the tip to the 3.5 cm dishes containing the pre-warmed growth medium (without antibiotics) (see Note 9).
- Gently move the dish horizontal and vertical to evenly distribute the cells (see Note 10).
- Incubate the cells in a humidified CO2 incubator (95% air, 5% CO2, 37 °C) for 48-72 h to allow efficient silencing (see Note 11).
- Discard the Neon tip in an appropriate biological hazardous waste container and repeat steps A12 to A18 for the remaining cell-DNA-siRNA mixtures (e.g., control siRNA and other plasmids of interest) (see Note 12).
- Perform all cell culture based work in a class II biological safety cabinet/tissue culture hood and disinfect the work surface and materials (e.g., pipettes) with 70% (v/v) ethanol.
- Live-cell imaging
- Prior to image acquisition set up a controlled-temperature and CO2 chamber at the microscope stage, as well as an objective warmer. In the absence of a CO2 regulator change cells to a CO2–independent medium (e.g., HEPES buffered).
- Set up a glass bottom dish in the controlled-temperature and CO2 chamber, switch on the mercury lamp and scan the sample for transfected cells (i.e., green GFP signal).
- Image collection is performed using VisiView software. For imaging, 250 stacks of 9 planes (0.5 µm thickness, 100 msec exposure) were taken from each cell in a continuous stream. All conditions and laser intensities were kept between experiments.
- Collected images are converted into a Maximum intensity projection file using VisiView. This option compresses the images from each stack into a single one and binds them in a continuous time frame (Videos 1 and 2).
 Video 1. Peroxisome movement in control fibroblasts. Video 1 shows movement of peroxisomes in human skin fibroblasts (control) transfected with the peroxisome marker EGFP-SKL (fluorescent reporter with a C-terminal peroxisomal targeting signal). Note the spherical, punctate labelling of peroxisomes and some long-range, directed movements, which are likely microtubule-dependent. Bar = 20 µm. Video 1 (2 min observation, 10 x original speed) is taken from Costello et al. (2017b). Video 2. Peroxisome movement in fibroblasts after loss of ACBD5. Video 2 shows increased movement of peroxisomes in human skin fibroblasts silenced for ACBD5. Peroxisomes were labelled as described (Video 1). Note that loss of ACBD5, which tethers peroxisomes to the ER, results in increased peroxisomal movement. Not all of the movements may depend on microtubules. Bar = 20 µm. Video 2 (2 min observation, 10 x original speed) is taken from Costello et al. (2017b).
Video 1. Peroxisome movement in control fibroblasts. Video 1 shows movement of peroxisomes in human skin fibroblasts (control) transfected with the peroxisome marker EGFP-SKL (fluorescent reporter with a C-terminal peroxisomal targeting signal). Note the spherical, punctate labelling of peroxisomes and some long-range, directed movements, which are likely microtubule-dependent. Bar = 20 µm. Video 1 (2 min observation, 10 x original speed) is taken from Costello et al. (2017b). Video 2. Peroxisome movement in fibroblasts after loss of ACBD5. Video 2 shows increased movement of peroxisomes in human skin fibroblasts silenced for ACBD5. Peroxisomes were labelled as described (Video 1). Note that loss of ACBD5, which tethers peroxisomes to the ER, results in increased peroxisomal movement. Not all of the movements may depend on microtubules. Bar = 20 µm. Video 2 (2 min observation, 10 x original speed) is taken from Costello et al. (2017b).
- Prior to image acquisition set up a controlled-temperature and CO2 chamber at the microscope stage, as well as an objective warmer. In the absence of a CO2 regulator change cells to a CO2–independent medium (e.g., HEPES buffered).
- Peroxisome motility measurementsThe following workflow is implemented as a Python module, which uses Numpy, Scipy, and Scikit-Image libraries.
- Each frame is filtered using Laplace of Gaussian scale-space filtering (Lindeberg, 1998).
- The threshold for each filtered image is determined using Median Absolute Deviation as a robust estimator of foreground locations (Hampel, 1974).
- Peroxisome positions are calculated as maxima in the filtered image above threshold.
- Positions between frames are tracked using a global optimization subroutine using a modified version of the Jonker-Volgenant algorithm (Jonker and Volgenant, 1987).
- Tracking results are manually verified for accuracy.
- Peroxisome trajectories are displayed as trajectory and density plots, and converted into speed distributions which are subsequently plotted as cumulative distribution functions (CDFs) (Figure 1).
- Pseudocode for the tracking is:
# Detection
allpositions = []
for t in range(len(frames)):
filtered = scale_space_filter(frames[t])
threshold = median(filtered) + 3*median_absolute_deviation(filtered)
positions = peak_locations(filtered, mask=filtered>threshold)
allpositions.append(positions)
# Tracking
allidentities = [ range(len(allpositions[0])) ]
max_identity = max(allidentites[0])
for t in range(1, len(allpositions)+1):
cost = distance_matrix(allpositions[t-1], allpositions[t])
identities, final_cost = lapjv(cost)
identities[final_cost > maximum_distance] = -1
max_identity = max(allidentites[-1], max_identity)
num_new = count(identities == -1)
identities[identities == -1] = range(max_identity+1, max_identity+num_new+1)
allidentities.append(identities)
# Create trajectories
for t in range(len(allidentities)):
for i in range(len(allidentities[t])):
trajectories[allidentities[t][i]].append(allpositions[t][i])
- Each frame is filtered using Laplace of Gaussian scale-space filtering (Lindeberg, 1998).
Data analysis
- Trajectory plots (Figures 1A-1C)
- Randomly select a fixed number of peroxisome trajectories for each condition being analysed–we used 100 trajectories.
- Smooth trajectories using Kalman filtering and an Expectation-Maximization algorithm (https://github.com/pykalman/pykalman: PyKalman: Kalman Filter, Smoother, and EM Algorithm for Python).
- Plot the initial 20 time frames of each trajectory, starting at the center (x = 0, y = 0), i.e., subtract the initial position from each trajectory before plotting (see Costello et al., 2017b).
- Randomly select a fixed number of peroxisome trajectories for each condition being analysed–we used 100 trajectories.
- Density plots (Figures 1D-1F)
- Select all significant trajectories–in our case we chose trajectories with 20 or more time frames.
- Smooth the trajectories using Kalman filtering and an Expectation-Maximization algorithm.
- Perform pooling and binning of the x and y coordinates of the trajectories; we used the interval -3.3 µm in x and y directions and 50 bins along each dimension.
- Plot the log-scaled 2D histogram of these points to accentuate the ‘wings’ of the distribution; we recommend a high contrast colormap, e.g., the ‘jet’ color-map available in Matplotlib (and MATLAB).
- Select all significant trajectories–in our case we chose trajectories with 20 or more time frames.
- ECDF plots (Figure 1G)
- Select all significant trajectories–in our case we chose trajectories with 20 or more time frames (see Costello et al., 2017b).
- Smooth the trajectories using Kalman filtering and an Expectation-Maximization algorithm.
- Calculate the instantaneous trajectory speed profiles by calculating the distance moved between each time point in the trajectory.
- Pool the speeds and convert them into an empirical cumulative distribution function (ECDF) by sorting the speeds.
- By pooling speeds for all datasets for a given condition, a single ECDF is generated for each (in our case we had a minimum of 38,175 trajectories from 24 videos per condition).
- The ECDF is generated using the following pseudocode:
xvals = sort(speeds)
yvals = range(N)/N
plot(xvals, yvals)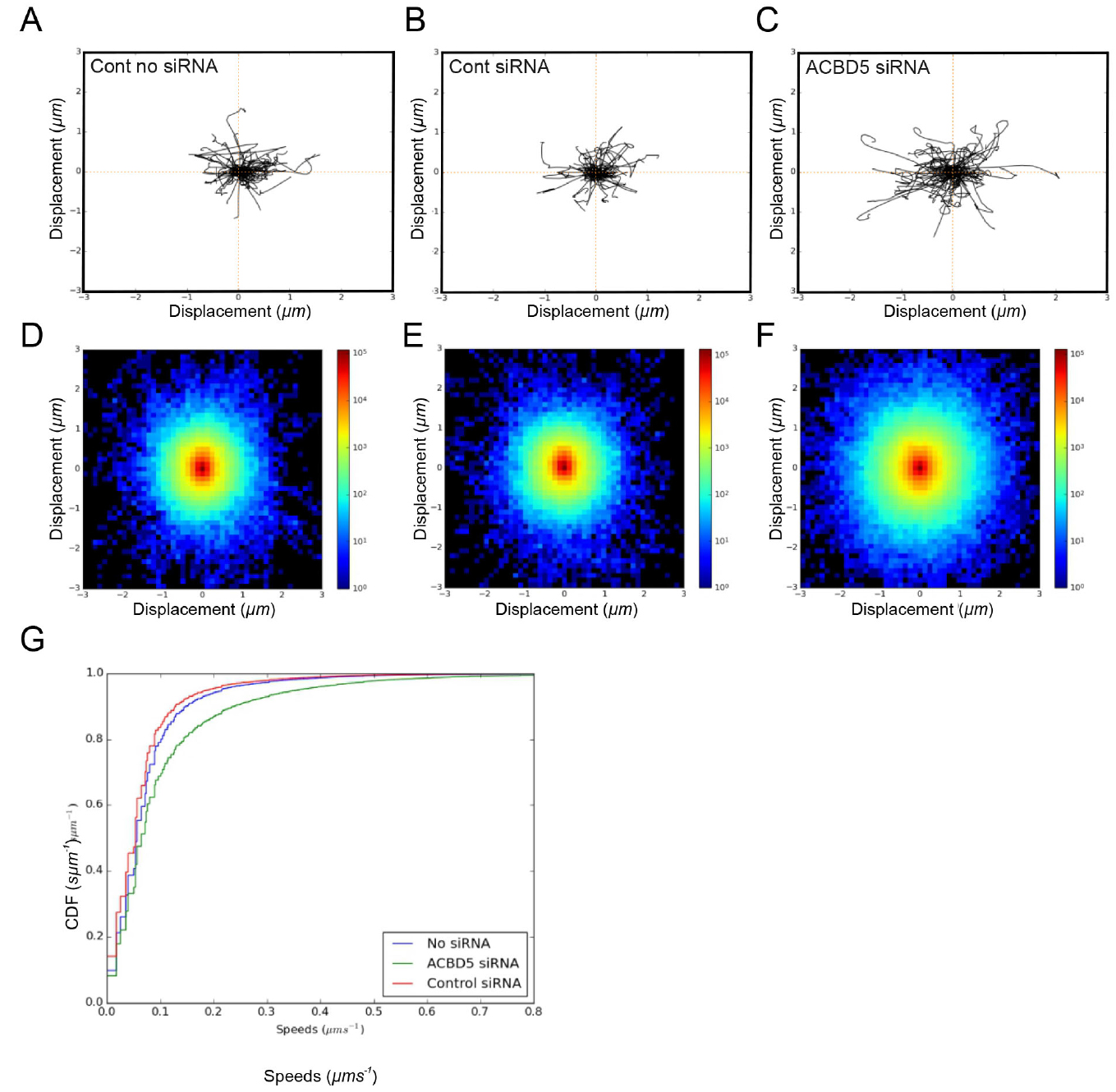
Figure 1. Analysis of peroxisome movement. A-C. For trajectory plots, 100 peroxisome trajectories were retrieved for each condition and the first 20 time-frames were plotted starting at a center. D-F. For density plots, the x and y coordinates of all trajectories ≥ 20 time-frames were pooled and binned in the interval -3,3 µm in x and y directions, using 50 bins. The log-scaled 2D histogram of these points was plotted using ‘jet’ colormap. G. For ECDF plots, instantaneous trajectory speed profiles were estimated by calculating distance moved between each time-point in the trajectory. These speeds were pooled and converted to an Empirical Cumulative Distribution Function (ECDF). By pooling speeds for all datasets for a given condition, a single ECDF was generated for each (min. 38,175 trajectories from 24 videos/condition). All three approaches for analysis of peroxisome motility revealed an increase in peroxisome movement under conditions where ACDB5, which is involved in the tethering of peroxisomes to the ER, is lost. Figure 1 is taken from Costello et al. (2017b).
- Select all significant trajectories–in our case we chose trajectories with 20 or more time frames (see Costello et al., 2017b).
Notes
- Other mammalian cell lines (e.g., COS-7 or HepG2 cells) can also be used. Those do not necessarily require microporation for transfection of plasmids or siRNA.
- Follow the biosafety and GMO guidelines of your institution.
- We usually pre-warm 1x PBS, TrypLETM Express solution, and complete growth medium to 37 °C. Medium is removed from the cell culture dish with a Pasteur pipette by vacuum aspiration. Cells are washed once with 3-4 ml of 1x PBS before TryplExpress solution is added (approx. 1 ml/10 cm dish) (gently tilt to cover the surface and incubate for 2-5 min at 37 °C). Upon detachment, cells are harvested in complete growth medium (10 ml/10 cm dish), carefully resuspended by pipetting the cell suspension 2-3 times up and down and transferred to a 15 ml conical tube. Cells are pelleted by centrifugation in a table top centrifuge (500 x g, 3 min at RT) and resuspended in 10 ml of complete growth medium. For maintenance, approx. 5 x 105 cells are transferred to a new 10 cm cell culture dish containing 10 ml of complete growth medium and incubated in a humidified CO2 incubator (95% air, 5% CO2, 37 °C).
- A detailed instruction manual of how to use the Neon® device can be found on the supplier’s website (http://tools.thermofisher.com/content/sfs/manuals/neon_device_man.pdf).
- Transfection conditions for other cell types need to be optimized. Further information is provided on the supplier’s website (https://www.thermofisher.com/be/en/home/life-science/cell-culture/transfection/transfection---selection-misc/neon-transfection-system/neon-protocols-cell-line-data.html).
- Cell number can also be determined manually, e.g., with a Neubauer chamber.
- It is advisable to collect a higher number of cells than required for each transfection (e.g., double the amount) to account for possible microporation failures due to air bubbles in the electroporation tip (visible electric discharge), and to facilitate Neon tip pipetting.
- If transfecting with more than one plasmid, pre-mix 1-2 µg of each plasmid.
- In the case of an electric spark, discard the tip with the cells and repeat the microporation using the backup cells.
- Do not swirl to avoid accumulation of the cells in the centre of the dish.
- The silencing efficiency should be tested and optimised prior to motility analysis by setting up similar dishes, and perform cell lysis and Western blotting. Efficient silencing can also be confirmed by immunofluorescence.
- The Neon pipette tips and tubes can be regenerated and reused to minimise costs of microporation (Brees and Fransen, 2014).
Recipes
- Complete growth medium for cell culture of human skin fibroblasts
Dulbecco’s modified Eagle medium (DMEM) high glucose (4.5 g/L), supplemented with:
10% fetal bovine serum (FBS)
100 U/ml of penicillin and 100 μg/ml of streptomycin
Store at 4 °C - Phosphate-buffered saline (1x PBS)
140 mM NaCl
2.5 mM KCl
6.5 mM Na2HPO4
1.5 mM K2HPO4, pH 7.35
Acknowledgments
We would like to thank J. Costello and T. Schrader for helpful comments and suggestions. This protocol was adapted from Costello et al. (2017b) J Cell Biol 216(2): 331-342. DOI: 10.1083/jcb.201607055. This work was supported by grants from the Biotechnology and Biological Sciences Research Council (BB/K006231/1 and BB/N01541X/1 to M. Schrader). J. Metz and M. Schrader are supported by a Wellcome Trust Institutional Strategic Support Award (WT097835MF and WT105618MA). M. Schrader is supported by Marie Curie Initial Training Network action PerFuMe (316723).
References
- Brees, C. and Fransen, M.(2014). A cost-effective approach tomicroporate mammalian cells with the Neon Transfection System. AnalBiochem 466: 49-50.
- Costello, J., Castro, I.,Camões, F., Schrader, T. A., McNeal, D., Gomes, S., Giannopoulo, N., Pogenberg,V., Bonekamp, N., Ribeiro, D., Wilmanns, M., Islinger, M., Schrader, M. (2017a). Predictingthe targeting of tail-anchored proteins to subcellular compartments inmammalian cells. J Cell Sci 130(9):1675-1687.
- Costello, J., Castro, I., Hacker, C., Schrader, T. A.,Metz, J., Zeuschner, D., Azadi, A. S., Godinho, L. F., Costina, V., Findeisen,P., Manner, A., Islinger, M., Schrader, M. (2017b). ACBD5 andVAPB mediate membrane associations between peroxisomes and the ER. J Cell Biol 216(2): 331-342.
- Costello, J., Castro, I.,Schrader, T. A., Islinger, M., Schrader, M. (2017c). Peroxisomal ACBD4interacts with VAPB and promotes ER-peroxisome associations. Cell Cycle 16(11): 1039-1045.
- Eisenberg-Bord, M., Shai, N.,Schuldiner, M. and Bohnert, M. (2016). A tether is a tether is atether: Tethering at membrane contact sites. DevCell 39(4): 395-409.
- Fagarasanu, A., Mast, F. D., Knoblach, B., Jin, Y., Brunner, M. J.,Logan, M. R., Glover, J. N., Eitzen, G. A., Aitchison, J. D., Weisman, L. S.,Rachubinski, R. A. (2009). Myosin-driven peroxisome partitioning in S. cerevisiae. J Cell Biol 186(4): 541-54.
- Ferdinandusse, S., Falkenberg,K. D., Koster, J., Mooyer, P. A., Jones, R., van Roermund, C. W. T., Pizzino,A., Schrader, M., Wanders, R. J. A., Vanderver, A. and Waterham, H. R. (2017). ACBD5deficiency causes a defect in peroxisomal very long-chain fatty acidmetabolism. J Med Genet 54(5):330-337.
- Guimaraes, S. C., Schuster,M., Bielska, E., Dagdas, G., Kilaru, S., Meadows, B. R., Schrader, M. andSteinberg, G. (2015). Peroxisomes, lipid droplets,and endoplasmic reticulum "hitchhike" on motile early endosomes. JCell Biol 211(5): 945-954.
- Hampel,F. R. (1974). The influence curve and its role in robust estimation. J Am Stat Assoc 69(346): 383-393.
- Hua, R., Cheng, D., Coyaud,E., Freeman, S., Di Pietro, E., Wang, Y., Vissa, A., Yip, C. M., Fairn, G. D.,Braverman, N., Brumell, J. H., Trimble, W. S., Raught, B. and Kim, P. K.(2017). VAPs and ACBD5 tetherperoxisomes to the ER for peroxisome maintenance and lipid homeostasis. JCell Biol 216(2): 367-377.
- Islinger, M., Cardoso, M. J.,Schrader, M. (2010). Be different–the diversityof peroxisomes in the animal kingdom. Biochem Biophys Acta 1803(8): 881-897.
- Islinger,M. and Schrader, M. (2011). Peroxisomes. Curr Biol 21(19): R800-801.
- Jedd,G. and Chua, N. H. (2002). Visualization of peroxisomesin living plant cells reveals acto-myosin-dependent cytoplasmic streaming andperoxisome budding. Plant Cell Physiol 43(4): 384-392.
- Jonker, R., and Volgenant, A.(1987). Ashortest augmenting path algorithm for dense and sparse linearassignment problems. Computing 38(4): 325-340.
- Lin, C., Schuster, M.,Guimaraes, S. C., Ashwin, P., Schrader, M., Metz, J., Hacker, C., Gurr, S. J.and Steinberg, G. (2016). Active diffusion andmicrotubule-based transport oppose myosin forces to position organelles incells. Nat Commun 7: 11814.
- Lindeberg, T. (1998). Feature detection with automatic scale selection. Int J Comput Vis 30(2): 79-116.
- Prinz, W. A. (2014). Bridgingthe gap: membrane contact sites in signaling, metabolism, and organelledynamics. J Cell Biol 205(6): 759-769.
- Rapp, S., Saffrich, R., Anton,M., Jakle, U., Ansorge, W., Gorgas, K. and Just, W. W. (1996). Microtubule-basedperoxisome movement. J Cell Sci 109 (Pt 4):837-849.
- Rodríguez-Serrano, M., Romero-Puertas, M. C., Sparkes, I., Hawes,C., del Rio, L. A. and Sandalio, L. M. (2009). Peroxisome dynamics in Arabidopsis plants under oxidativestress induced by cadmium. Free Radic Biol Med 47(11): 1632-1639.
- Schrader, M., Burkhardt, J.K., Baumgart, E., Luers, G., Spring, H., Volkl, A. and Fahimi, H. D. (1996). Interactionof microtubules with peroxisomes. Tubular and spherical peroxisomes in HepG2cells and their alterations induced by microtubule-active drugs. EurJ Cell Biol 69(1): 24-35.
- Schrader, M., Godinho, L. F.,Costello, J. L. and Islinger, M. (2015). The different facets oforganelle interplay-an overview of organelle interactions. FrontCell Dev Biol 3: 56.
- Schrader, M., Grille, S.,Fahimi, H. D. and Islinger, M. (2013). Peroxisome interactions andcross-talk with other subcellular compartments in animal cells. SubcellBiochem 69: 1-22.
- Schrader, M., King, S. J.,Stroh, T. A. and Schroer, T. A. (2000). Real time imaging reveals aperoxisomal reticulum in living cells. JCell Sci 113 (Pt 20): 3663-3671.
- Schrader, M., Thiemann, M. andFahimi, H. D. (2003). Peroxisomal motility andinteraction with microtubules. MicroscRes Tech 61(2): 171-178.
- Wanders, R. J., Waterham, H.R. and Ferdinandusse, S. (2015). Metabolicinterplay between peroxisomes and other subcellular organelles includingmitochondria and the endoplasmic reticulum. FrontCell Dev Biol 3: 83.
- Waterham, H. R.,Ferdinandusse, S. and Wanders, R. J. (2016). Human disorders of peroxisomemetabolism and biogenesis. Biochim Biophys Acta 1863(5): 922-933.
- Wiemer, E. A., Wenzel, T.,Deerinck, T. J., Ellisman, M. H. and Subramani, S. (1997). Visualizationof the peroxisomal compartment in living mammalian cells: dynamic behavior andassociation with microtubules. JCell Biol 136(1): 71-80.
- Yagita,Y., Shinohara, K., Abe, Y., Nakagawa, K., Al-Owain, M., Alkuraya, F. S. andFujiki, Y. (2017). Deficiency of a retinaldystrophy protein, acyl-CoA bindingdomain-containing 5 (ACBD5), impairs peroxisomal β-oxidation of very-long-chainfatty acids. J Biol Chem 292(2):691-705.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Metz, J., Castro, I. G. and Schrader, M. (2017). Peroxisome Motility Measurement and Quantification Assay. Bio-protocol 7(17): e2536. DOI: 10.21769/BioProtoc.2536.
Category
Cell Biology > Cell imaging > Live-cell imaging
Cell Biology > Cell-based analysis > Organelle motility
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.