- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Dense sgRNA Library Construction Using a Molecular Chipper Approach
Published: Vol 7, Iss 12, Jun 20, 2017 DOI: 10.21769/BioProtoc.2373 Views: 10926
Reviewed by: Gal HaimovichKabin XieAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Genetic screens using single-guide-RNA (sgRNA) libraries and CRISPR technology have been powerful to identify genetic regulators for both coding and noncoding regions of the genome. Interrogating functional elements in noncoding regions requires sgRNA libraries that are densely covering, and ideally inexpensive, easy to implement and flexible for customization. We present a Molecular Chipper protocol for generating dense sgRNA libraries from genomic regions of interest. This approach utilizes a combination of random fragmentation and a Type III restriction enzyme to derive a dense coverage of sgRNA library from input DNA.
Keywords: Molecular chipperBackground
Genome editing using Streptococcus pyogenes (sp) Cas9 and sgRNA libraries is a powerful tool to screen for functional genetic regulators in mammalian cells by generating biallelic loss-of-function sequence alterations (Wiedenheft et al., 2012; Mali et al., 2013; Koike-Yusa et al., 2014; Shalem et al., 2014; Wang et al., 2014; Zhou et al., 2014). Cas9 binds sgRNA, which can be designed to target Cas9 toward a defined locus in the genome. The nuclease activity of Cas9 cuts target DNA locus, leading to double-stranded DNA breaks, which upon DNA repair through non-homologous end-joining pathway frequently results in short deletions at the locus of interest.
The powerful genomic editing capacity of the CRISPR-Cas9 system has led to the use of sgRNA libraries to interrogate protein-coding genes as well as noncoding regions. Several sgRNA libraries for protein-coding genes and/or limited numbers of non-coding genes have been reported in functional screening, through sgRNA enrichment, to identify genes and networks regulating specific cellular functions (Koike-Yusa et al., 2014; Shalem et al., 2014; Wang et al., 2014; Zhou et al., 2014; Canver et al., 2015; Sanjana, 2016). Several non-coding sgRNA libraries consisting of 703-18,000 sgRNAs densely covering regulatory regions of genes of interest, such as BCL11A, Tdgf1a and drug-resistance regulating genes, were also reported in gene-specific functional screens for distal and proximal regulating elements (Canver et al., 2015; Rajagopal et al., 2016; Korkmaz et al., 2016; Sanjana, 2016). These sgRNA libraries were all produced by careful bioinformatics design, oligonucleotide synthesis on microarray, and cloning of oligonucleotide pool(s) into vectors. This synthetic approach has been very useful, but requires computational expertise for genome-wide sgRNA design and expensive microarray synthesis, and thus is challenging for most laboratories.
Enzymatically generated sgRNA libraries covering regions of repetitive genomic sequences or loci are useful for CRISPR-Cas9 imaging of genomic sequences or loci (Lane et al., 2015). Due to lack of high-density (~111 bp), such sgRNA libraries are not reported in screening for functional non-coding regions. Another enzymatic method was reported to generate high-density (~20 bp) sgRNA library from cDNA (Arakawa, 2016). This type of sgRNA library consists of cell source-specific, differentially expressed sequences, thus, was neither reported for applications in functional screening.
Without prior knowledge of the locations of critical noncoding-element-containing regions, functional mapping of noncoding genomic regions requires sgRNA libraries that densely populate regions of interest. The ideal method requires flexibility for adjusting the scale of sgRNA production to easily cope with this need. We describe here a detailed protocol of the Molecular Chipper approach that processes any input DNA piece(s) to generate a near base-resolution sgRNA library densely covering the input DNA of interest.
Materials and Reagents
- Pipette tips (prefer ones with filters to minimize contamination, such as those from Denville Scientific)
- Tubes (Denville Scientific, catalog number: C2170 )
- Petri dishes (Corning, Falcon®, catalog number: 351029 )
- NEB 5-alpha Electrocompetent E. coli (New England Biolabs, catalog number: C2989K )
- Retroviral vector pSUPER-CRISPR that contains and a puromycin selection marker and a U6 promoter to drive expression of sgRNA that is cloned at BamHI-HindIII sites (for details, see Cheng et al., 2016b).
Note: Please write us to request for this material. - T4 DNA ligase at 2000,000 U/ml (New England Biolabs, catalog number: M0202T ). Use in ligations where T4 DNA ligase is required in excess within a small volume, such as that described in step 2
- T4 DNA ligase at 400,000 U/ml (New England Biolabs, catalog number: M0202S )
- T4 polynucleotide kinase (New England Biolabs, catalog number: M0201S )
- Distilled water (AmericanBio, catalog number: AB02123-00500 )
- QIAquick PCR Purification Kit (QIAGEN, catalog number: 28104 )
- Agarose (AmericanBio, catalog number: AB00972 )
- Ethidium bromide, 10 mg/ml (Sigma-Aldrich, catalog number: E1510 )
- 3 M sodium acetate, pH 5.2
- 100% ethanol (AmericanBio, catalog number: AB00515-00500 )
- 70% ethanol (AmericanBio, catalog number: AB04010-00500 )
- 1 kb plus DNA standard (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10787018 )
- Agarose, low melting point (AmericanBio, catalog number: AB00981 )
- 10 bp DNA standard (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10821015 )
- NEBNext End Repair Module (New England Biolabs, catalog number: E6050S )
- 10,000x SYBR Safe DNA Gel Stain (Thermo Fisher Scientific, InvitrogenTM, catalog number: S33102 )
- QIAEX II Gel Extraction Kit (QIAGEN, catalog number: 20021 )
- QIAquick Gel Extraction Kit (QIAGEN, catalog number: 28704 )
- EcoP15I-adaptor oligonucleotide pair: sense aaaactcgagcagcagtggatccG and anti-sense /5phos/Cggatccactgctgctcgag (Integrated DNA Technologies; 25 nmole DNA oligo scale; Standard desalting purification). The anti-sense oligo has a 5’-phosphase modification
- 100 bp DNA standard (New England Biolabs, catalog number: N3231S )
- EcoP15I enzyme (New England Biolabs, catalog number: R0646L )
- PCI: Phenol:Chloroform:Isoamyl Alcohol 25:24:1, saturated with TE (10 mM Tris, pH 8.0, 1 mM EDTA) (Sigma-Aldrich, catalog number: P2069-100ML )
- Phenol, saturated with Tris, pH 7.5 (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 17914 )
- Chloroform (AmericanBio, catalog number: AB00350-00500 )
- PCI (Phenol:chloroform:isoamyl alcohol 25:24:1), Tris saturated (Roche Diagnostics, catalog number: 03117944001 )
- 3’ sgRNA backbone adaptor oligo nucleotide pair: sense /5phos/nngttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgc-tttttttaagctttat and anti-sense ataaagcttaaaaaaagcaccgactcggtgccactttttcaagttgataac-ggactagccttattttaacttgctatttctagctctaaaac (Integrated DNA Technologies, 100 nmole DNA oligo scale, Standard desalting purification). The -sense oligo has a 5’-phosphase modification
- BamHI-HF enzyme (New England Biolabs, catalog number: R3136S )
- HindIII-HF enzyme (New England Biolabs, catalog number: R3104S )
- MiniElute Gel Extraction kit (QIAGEN, catalog number: 28604 )
- LB medium (Thermo Fisher Scientific, InvitrogenTM, catalog number: 12795027 )
- QIAprep Spin Miniprep Kit (QIAGEN, catalog number: 27104 )
- Oligo nucleotide used in Sanger sequencing of cloned-sgRNA: ctccctttatccagccctca (Intergatred DNA Technologies, 25 nmole DNA oligo scale, Standard desalting purification)
- Ampicillin (AmericanBio, catalog number: AB00115-00100 )
- Tris base (Sigma-Aldrich, catalog number: T6066 )
- Ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich, catalog number: EDS-100G )
- Glacial acetic acid (Sigma-Aldrich, catalog number: 695092 )
- Agar (AmericanBio, catalog number: AB01185-00500 )
- 50x TAE gel running buffer (see Recipes)
Equipment
- Pipettes
- 37 °C water bath (Fisher Scientific, model: Model 215 , catalog number: 15-462-15Q)
- NanoDrop 2000 (Thermo Fisher Scientific, model: NanoDropTM 2000 , catalog number: ND-2000)
- Microcentrifuge (Eppendorf, model: 5254 , catalog number: 022620444)
- S220 Focused-ultrasonicator and sonication vials (COVARIS, model: S220 )
- Gel Illuminator (UltraSlim LED Illuminator, Maestrogen, catalog number: SLB-01W )
- IncuBlock heating block (Danville Scientific, model: I-0259 , catalog number: 08302)
- Gel electrophoresis system (Thermo Fisher Scientific, Thermo ScientificTM, model: OwlTM EasyCastTM B1A , catalog number: B1A)
- Electroporation System (Bio-Rad Laboratories, model: Gene Pulser XcellTM, catalog number: 1652660 )
Procedure
- For large DNA fragments, such as BAC clones, start from step 6. If input DNA is small and composed of multiple pieces, such as individual PCR products, purified DNA pieces are quantified by a NanoDrop 2000 and pooled in an equal molar ratio.
- If using an input pool of multiple pieces of DNA, randomly ligated larger products are then generated using T4 DNA ligase in excess. Specifically, for each μg of DNA, the DNA pool are ligated with 4,000 U of T4 DNA ligase (New England Biolabs) in a 50 μl reaction for 3 h at 37 °C. We start with ~20 μg total DNA that has 5’-phosphate groups.
Note: This step has been added to avoid biasing against regions located near the ends of input DNA pieces (because we perform a size selection after this fragmentation step, and sequences close to the ends would be represented by very small fragments after fragmentation, and thus would be under-represented in the final library). - If directly using PCR products, perform a kinase reaction prior to ligation using T4 polynucleotide kinase (TPK). Specifically, we followed the manufacture NEB’s protocol of a reaction consisting of 300 pmol of PCR DNA, 5 μl of 10x buffer, 5 μl of 10 mM ATP, and 1 μl (10 U) of T4 PNK, supplemented with distilled water to total 50 μl, The reaction is incubated at 37 °C for 30 min, followed by inactivation at 65 °C for 20 min. DNA in the phosphorylation reaction is then purified by QIAGEN PCR DNA purification kit and eluted in 50 μl of distilled water prior to ligation.
- Run 10 μl or 200 ng of ligation products on 1% agarose gel with 0.5 μg/ml ethidium bromide in 1x TAE buffer, in order to check sizes of ligation products (usually > 10 kb on average).
- The ligated large amount of DNA is purified by ethanol precipitation. Add 10% volume of 3 M sodium acetate, pH 5.2, and 2 volumes of 100% ethanol; mix and then precipitate in -20 °C for one hour; spin down using a pre-cooled microcentrifuge for 10 min, wash with 70% ethanol; air dry for one minute; and re-suspend in 150 μl water.
- To generate random DNA fragments, ~14 μg of the input DNA (ligated if originating from multiple pieces) in 120 μl of water is sonicated in a S220 Focused-ultrasonicator for 90 sec to result in fragments peaking at sizes of 400-450 bp (Peak Power = 140 V, Duty Factor = 5, Cycle/Burst = 200 and Average Power = 7). Run at least 2-4 μl (250-500 ng) DNA using 1 kb Plus DNA standard (Invitrogen) on 2% agarose gel with ethidium bromide to check peak sizes of fragmented DNA (Figure 1A).
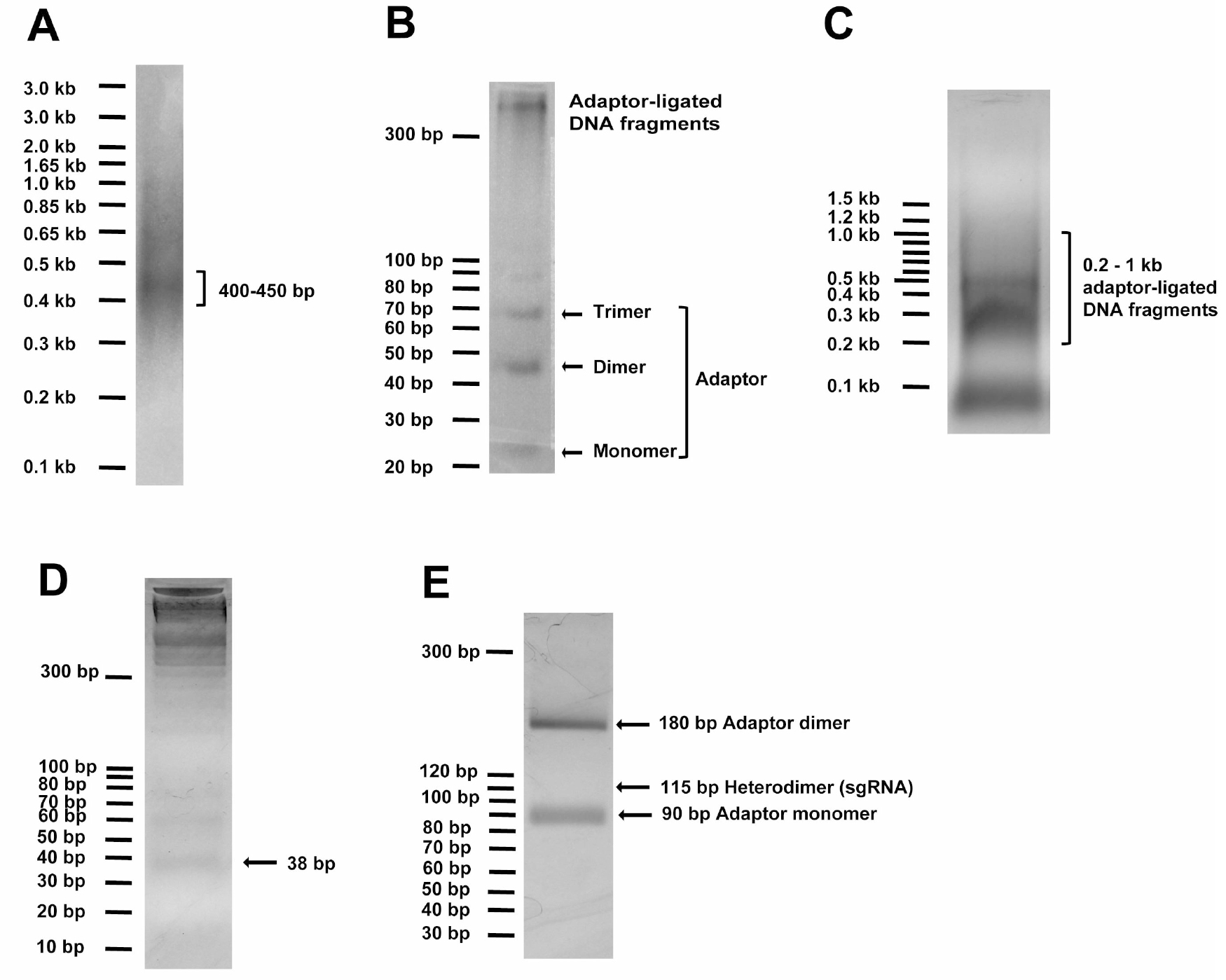
Figure 1. DNA gel examples of critical steps in Molecular Chipper procedure. A. Sonicated DNA (2-4 μl or 250-500 ng) in step 6 was visualized on 2% agarose gel with the help of 250 ng of 1 kb Plus DNA standard, showing peak fragment size of 400-450 bp. B. 5 μl (~200 ng) of DNA fragments ligated to adaptor in step 9 was visualized on a 4% LMP agarose gel with 10 bp DNA standard, showing formation of adaptor dimer and trimer and ligated DNA fragment at higher molecular weight. C. The total ligation in step 9 was loaded onto a 1% agarose gel to visualize and to recover ligated DNA fragments in 200-1,000 bp linear DNA range, as detailed in step 10. D. Adaptor-ligated DNA fragments were digested with EcoP15I to release the 38-bp DNA ends and loaded onto 4% LMP agarose gel to purify the 38-bp DNA pool, as detailed in step 11. E. The full-length sgRNA pool digested with BamHI and HindIII was loaded onto 4% LMP agarose gel to view and purify the 115-bp sgRNA DNA pool, as detailed in step 15.
Note: the ~115-bp DNA band appears faint due to high background. - Sonicated fragments are repaired in a 150 μl End Repair reaction with 15 μl of the NEBnext End Repairing Enzyme Mix, 30 μl of NEBNext End Repair Reaction 10x buffer, supplemented with distilled water to total 300 μl and react at 25 °C for 30 min, followed by 1% agarose gel purification of the 400-450 bp DNA fragments (visualized with 1x SYBR Safe in the gel by a blue-light gel dock, such as the Maestrogen Illuminator, in order to avoid DNA damage under UV light especially short-wave UV light that is usually available in labs, when ethidium bromide is used to visualize DNA) by using QIAEX II Gel Extraction Kit.
- To obtain fragment ends from both ends of the random DNA fragments, an EcoP15I-adaptor is first prepared by annealing two oligonucleotides aaaactcgagcagcagtggatccG and /5phos/Cggatccactgctgctcgag (IDT) in equal molar ratio in 1x ligation buffer (NEB) at 10 μM by heating up to 95 °C and then gradually decreasing temperature by 1 °C per minute to 25 °C in a PCR machine or by incubating in boiled water cooling down gradually to room temperature. The annealed DNA adaptor contains an EcoP15I site (in bold) followed by a total 8-bp spacer, including a BamHI site (underlined) and a G (capitalized) at the end for later sgRNA cloning to serve as transcription start site from the U6 promoter.
- ~12 μg of the DNA fragments are ligated, at a ~1:10 molar ratio to 6.0 μg of the above annealed EcoP15I-adaptor with 20,000 U T4 DNA ligase (New England Biolabs) in 300 μl reaction for 3 h at 37 °C. Run 5 μl of ligation on 4% low melting point (LMP) gel with 10-bp DNA standard (Invitrogen) to confirm successful ligation by visualizing formation of 44-bp adaptor dimer (we also saw trimer formation) and the presence of higher MW DNA (Figure 1B).
- The adaptor-ligated DNA fragments in 200-1,000-bp linear standard range are purified from adaptor monomer and other non-specific bands by running them on a 1% agarose gel with NEB 100 bp DNA ladder (Figure 1C) and by using QIAEX II Gel Extraction Kit. 1% gel is important to minimize gel volume to increase yield.
- ~5 μg of the EcoP15I-adaptor-ligated gel-purified DNA is digested by 100 U of EcoP15I enzyme (New England Biolabs) in 300 μl reaction with specified buffer and ATP concentration for exactly 1 h at 37 °C. To check completeness of digestion and efficiency of adaptor ligation in step 9, run 15 μl of the digestion with 10 bp DNA standard (Invitrogen) on 4% low-melting-point agarose gel with ethidium bromide to visualize the 38 bp (expecting ~25-50 ng of the 38 bp DNA fragment; Figure 1D).
- After digestion, EcoP15I digestion reaction is cleaned by equal-volume sequential phenol/PCI/chloroform extractions and centrifugation at 18,400 x g for 5 min after each extraction, to get rid of EcoP15I protein bound to DNA potentially. The upper phase is always collected during repeated extraction. The final supernatant is precipitated with ethanol by adding 1/10 volume of 3 M sodium acetate, pH 5.7, and 2-volume of 100% ethanol to precipitate DNA, centrifuging at 18,400 x g for 10 min and washing DNA pellet with 70% ethanol, air-dried and finally re-suspended the DNA pellet in 50 μl of distilled water.
- Precipitated digestion products are gel-purified (on 4% low-melting-point agarose gel) to obtain a ~38-bp DNA fragment pool (EcoP15I-adaptor + 19/17 bases from ends of random DNA fragments) by diluting the gel slice with 2 volumes of 1x TAE buffer, melting the gel slice at 70 °C on a heating block, extracting by phenol/PCI/chloroform and dissolving DNA in 20 μl water.
- To ligate to the rest of sgRNA backbone, 280 ng of the purified 38-bp DNA pool is ligated in 50 μl reaction with 4,000 U (New England Biolabs, M0202T) of T4 DNA ligase for 3 h at 37 °C, at a 1:5 molar ratio to 2.75 μg of a sgRNA-backbone-adaptor. The sgRNA-backbone-adaptor contains two Ns for binding to overhangs from EcoP15I digestion products, the remaining sgRNA sequence (without the target-recognition domain), a polyT stretch for polymerase III transcriptional termination, and a HindIII site for cloning. This sgRNA-backbone adaptor was prepared by annealing two oligonucleotides below, followed by 4% low-melting-point agarose gel-purification, by using QIAEX II Gel Extraction Kit, to eliminate improperly annealed products.
Two oligonucleotides: /5phos/nngttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgc-tttttttaagctttat
ataaagcttaaaaaaagcaccgactcggtgccactttttcaagttgataacggactagccttattttaacttgctatttctagctctaaaac (IDT).
See step 8 for details of annealing the two oligonucleotides to prepare for the sgRNA-backbone adaptor DNA. - The ligated sgRNA DNA pool is cleaned by QIAquick PCR Purification Kit, digested in 50 μl with 20 U each of BamHI and HindIII (New England Biolabs) overnight at 37 °C, and gel-purified by running with 10 bp DNA standard (Invitrogen) on 4% low-melting-point agarose (Figure 1E) and by using the MiniElute Gel Extraction Kit to obtain a ~115-bp sgRNA pool. The 115-bp DNA band on gel usually appears faint due to high gel background when running large ligated DNA amount.
- This sgRNA pool is quantified by SYBR Safe Gel Stain (Invitrogen) on a fluorometer (total ~10 ng sgRNA pool DNA generated from starting material specified in step 9), and ligated at 3:1 molar ratio with 40 U of T4 DNA ligase (New England Biolabs) per 100 ng total DNA per 10 μl reaction at room temperature overnight into BamHI-HindIII sites of a retroviral vector pSUPER-CRISPR which contains a U6 promoter and a puromycin selection marker (Cheng et al., 2016b).
- The ligated DNA is ethanol precipitated (see step 4 for details) and dissolved in 10 μl water.
- Ligation products are transformed into NEB5 alpha competent cells in 30 μl aliquots by electroporation using a GenePulser Xcell. The transformed cells from 10 electro-transformations are then pooled together, grown in 10-ml LB medium for 1 h. Several small fractions of transformation are plated to estimate total transformed clones. To estimate the % cloning efficiency, 10-20 colonies are grown up to miniprep DNA using the QIAprep Spin Miniprep Kit, and the cloned sgRNA inserts are confirmed by Sanger sequencing using oligo nucleotide ctccctttatccagccctca. We found that about 80% clones have 19-20 mer sgRNAs cloned. The other 20% clones have much shorter sgRNAs cloned, likely due to non-specific cleavage activity of the EcoP15I enzyme.
- The transformation culture is grown overnight in 100 ml of LB medium containing 100 μg ml-1 of ampicillin for plasmid DNA preparation. Properly prepared, a library of > 1 million clones can be easily achieved.
- For retrovirus preparation, an example of functional reporter screen, sgRNA enrichment data analysis to identify functional non-coding genomic regions, and examples of mechanistic studies, see our previous publication (Cheng et al., 2016b).
Data analysis
- Sanger DNA sequencing data was analyzed using the DNA alignment feature in the ApE program, which can be downloaded and installed free at http://biologylabs.utah.edu/jorgensen/wayned/ape/. To do that, open the two sequences in ApE, choose Align Two Sequences under Tools in the program, specify the two sequence files in the pull-down menu in the new window, and choose OK to compute and generate the new alignment file.
- sgRNA library complexity was calculated as total E. coli colony number on plate times dilution factors.
Notes
- To better understand different stages of the construction of the sgRNA library, see a flow chart of the entire technical procedure in our previous publication (Cheng et al., 2016b) and a general conceptual flow chart of Molecular chipper-sgRNA library generation in comparison to conventional array-synthesized sgRNA library generation (Cheng et al., 2016a).
- The specified, optimized ligation and digestion time is important.
- Starting with a large amount of input DNA is critical. We start with minimal 20 μg of input DNA in our original report (Cheng et al., 2016b) and we usually prepare at least 40-60 μg of starting DNA material in case of failure at any intermediate step in the procedure.
- 1:5 molar ratio ligation to adaptors is important for efficient ligation of adaptors to both ends of DNA fragments. More adaptors may help to increase the ligation efficiency, but would cause higher background in gel purification, thus difficult to visualize the ligated DNA products.
- Obtaining hundreds of nanograms of the 38 bp fragments for ligating to the sgRNA backbone is a must for going forward.
- Desired complexity/coverage is achieved by transforming enough high-competency competent cells. We compared available commercial cells and found that NEB 5-alpha Electrocompetent E. coli is among the highest competency.
- If there are no 38 bp bands present after the first EcoP15I digestion, 1) Check if the adaptor has annealed well: ligate only the adaptors which should result in the presence of a major band of the monomer and minor non-specific bands on 4% low melting point gel; 2) Check if sonicated DNA are repaired well and if ligation goes well: treat a small fraction of repaired DNA by Taq DNA polymerase in the presence of dNTPs at 72 °C for 5 min, followed by TA cloning (Invitrogen): and many white colonies should appear if end repairing works well and sequencing of the cloned inserts should reveal adaptors at both ends of the cloned fragments; 3) Purify the adaptors from impurities by running on 3-4% LMP gel to reduce background during gel purification of the ligated DNA.
- If there are not enough colonies/coverage after transforming the final ligation, 1) Identify a commercial source of competent cell with high transformation efficiency, e.g., electroporation competent cells from NEB; 2) Alternatively, make your own electro-competent cells of higher efficiency; 3) Perform a test transformation to calculate total colony number/complexity/coverage before transforming the rest of ligation to make the whole DNA library.
- If cloning efficiency is not high, 1) Digest the full-length ligated sgRNA fragment with BamHI and HindIII for 6 h instead of overnight to minimize star activity; 2) Test the vector quality by cloning a mock BamHI-HindIII fragment and if necessary, a stuffer can be cloned first for efficient preparation of the double-enzyme digested vector.
Recipes
- 50x TAE gel running buffer per liter
242 g Tris base
18.61 g EDTA
57.1 ml glacial acetic acid
Dissolve in distilled water and bring final volume to 1 L
Acknowledgments
This study was supported in part by NIH grants R01CA149109 (to J.L.) and R01GM099811 (to Y.D. and J.L.). The protocol was adapted from previous work (Cheng et al., 2016a and 2016b). The authors declare no conflicts of interest or competing interests that may impact the design and implementation of this protocol.
References
- Arakawa, H. (2016). A method to convert mRNA into a gRNA library for CRISPR/Cas9 editing of any organism. Sci Adv 2(8): e1600699.
- Canver, M. C., Smith, E. C., Sher, F., Pinello, L., Sanjana, N. E., Shalem, O., Chen, D. D., Schupp, P. G., Vinjamur, D. S., Garcia, S. P., Luc, S., Kurita, R., Nakamura, Y., Fujiwara, Y., Maeda, T., Yuan, G. C., Zhang, F., Orkin, S. H. and Bauer, D. E. (2015). BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527(7577): 192-197.
- Cheng, J., Pan, W., Roden, C., Chen, Z., and Lu, J. (2016a). Molecular Chipper: Functional Mapping of the Non-Coding Genome with CRISPR. Next Generat Sequenc & Applic 3: 132.
- Cheng, J., Roden, C. A., Pan, W., Zhu, S., Baccei, A., Pan, X., Jiang, T., Kluger, Y., Weissman, S. M., Guo, S., Flavell, R. A., Ding, Y. and Lu, J. (2016b). A Molecular Chipper technology for CRISPR sgRNA library generation and functional mapping of noncoding regions. Nat Commun 7: 11178.
- Koike-Yusa, H., Li, Y., Tan, E. P., Velasco-Herrera Mdel, C. and Yusa, K. (2014). Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol 32(3): 267-273.
- Korkmaz, G., Lopes, R., Ugalde, A. P., Nevedomskaya, E., Han, R., Myacheva, K., Zwart, W., Elkon, R. and Agami, R. (2016). Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat Biotechnol 34(2): 192-198.
- Lane, A. B., Strzelecka, M., Ettinger, A., Grenfell, A. W., Wittmann, T. and Heald, R. (2015). Enzymatically generated CRISPR libraries for genome labeling and screening. Dev Cell 34(3): 373-378.
- Mali, P., Esvelt, K. M. and Church, G. M. (2013). Cas9 as a versatile tool for engineering biology. Nature methods 10(10): 957-963.
- Rajagopal, N., Srinivasan, S., Kooshesh, K., Guo, Y., Edwards, M. D., Banerjee, B., Syed, T., Emons, B. J., Gifford, D. K. and Sherwood, R. I. (2016). High-throughput mapping of regulatory DNA. Nat Biotechnol 34(2): 167-174.
- Sanjana, N. E. (2016). Genome-scale CRISPR pooled screens. Anal Biochem.
- Shalem, O., Sanjana, N. E., Hartenian, E., Shi, X., Scott, D. A., Mikkelsen, T. S., Heckl, D., Ebert, B. L., Root, D. E., Doench, J. G. and Zhang, F. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343(6166): 84-87.
- Wang, T., Wei, J. J., Sabatini, D. M. and Lander, E. S. (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science 343(6166): 80-84.
- Wiedenheft, B., Sternberg, S. H. and Doudna, J. A. (2012). RNA-guided genetic silencing systems in bacteria and archaea. Nature 482(7385): 331-338.
- Zhou, Y., Zhu, S., Cai, C., Yuan, P., Li, C., Huang, Y. and Wei, W. (2014). High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 509(7501): 487-491.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Cheng, J., Pan, W. and Lu, J. (2017). Dense sgRNA Library Construction Using a Molecular Chipper Approach. Bio-protocol 7(12): e2373. DOI: 10.21769/BioProtoc.2373.
Category
Molecular Biology > DNA > DNA cloning
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.