- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Targeted Mutagenesis Using RNA-guided Endonucleases in Mosses
Published: Vol 7, Iss 12, Jun 20, 2017 DOI: 10.21769/BioProtoc.2359 Views: 13349
Reviewed by: Dennis NürnbergVinay PanwarAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
RNA-guided endonucleases (RGENs) have been used for genome editing in various organisms. Here, we demonstrate a simple method for performing targeted mutagenesis and genotyping in a model moss species, Physcomitrella patens, using RGENs. We also performed targeted mutagenesis in a non-model moss, Scopelophilla cataractae, using a similar method (Nomura et al., 2016), indicating that this experimental system could be applied to a wide range of mosses species.
Keywords: Genome editingBackground
Targeted mutagenesis using RNA-guided endonucleases (RGENs) derived from the adaptive immune system, using the bacterial CRISPR (clusters of regularly interspaced palindromic repeats)/Cas (CRISPR-associated) systems, has dramatically advanced in recent years. In this method, the Cas9 endonuclease, derived from Streptococcus pyogenes, and an artificially designed single-chain guide RNA (sgRNA) are used. The Cas9-sgRNA complex recognizes the protospacer-adjacent motif (5’-NGG-3’) and cleaves 3 bp upstream of the target site (Jinek et al., 2012). Subsequently, random insertion and/or deletion mutations occur during the repair process for double-strand breaks (DSBs) in the DNA. As targeted mutagenesis using these RGENs is efficient as well as cost- and time-effective, it has been used for genome editing in various organisms, including many plant species. Here, we established a protocol for targeted mutagenesis using RGENs in mosses, and demonstrated it in a model and a non-model species (Nomura et al., 2016).
Materials and Reagents
- Washed and autoclaved cellophane (FUTAMURA CHEMICAL, catalog number: PS-1 )
- 9 cm plastic Petri dish (SANSEI MEDICAL, catalog number: 01-013 )
- 1.5 ml plastic tubes (FUKAEKASEI and WATSON, catalog number: 131-415C )
- 15 ml plastic tubes (FUKAEKASEI and WATSON, catalog number: 1332-015S )
- PCR tubes (NIPPON Genetics, FastGene, catalog number: FG-028DC )
- 10 µl plastic pipette tips (FUKAEKASEI and WATSON, catalog number: 123R-254CS )
- 200 µl plastic pipette tips (FUKAEKASEI and WATSON, catalog number: 123R-755CS )
- 1,000 µl plastic pipette tips (FUKAEKASEI and WATSON, catalog number: 122-804B )
- Protonemata of Physcomitrella patens (or other mosses)
- pSCgRNA, pSCOE1-fcoCas9 (Nomura et al., 2016, Figure 1A)
- DH5α-competent cells (Home-made)
- NEBuffer 2.1 (New England Biolabs, catalog number: R0539S )
- BbsI (New England Biolabs, catalog number: R0539S )
- Sterile Milli-Q water
- GEL/PCR Purification Mini Kit (Favorgen Biotech, catalog number: FAGCK 001-1 )
- Ligation high ver.2 (TOYOBO, catalog number: LGK-201 )
- ScU6p Ins. check F primer 5’-GAGGATCACGGTGTCACATGTCC-3’
- Quick Taq® HS DyeMix (TOYOBO, catalog number: DTM-101 )
- ScU6p Seq. check F primer 5’-ATGTCAAACATAACCTGG-3’
- Ampicillin (NACALAI TESQUE, catalog number: 02739-74 )
- Plasmid DNA Extraction Mini Kit (Favorgen Biotech, catalog number: FAPDE 001-1 )
- Agarose S (NIPPON GENE, catalog number: 312-01193 )
- DNA ladder markers (SMOBIO Technology, catalog number: DM3100 )
- NucleoBond® Xtra Midi (MACHEREY-NAGEL, catalog number: 740410.50 )
- G418 Disulfate (NACALAI TESQUE, catalog number: 16512-81 )
- BCDAT medium (Nishiyama et al., 2000)
- Glucose (Wako Pure Chemical Industries, catalog number: 049-31165 )
- Tks Gflex DNA polymerase (Takara Bio, catalog number: R060A )
- Zero Blunt PCR Cloning Kit (Thermo Fisher Scientific, Invitrogen, catalog number: K275020 )
- CloneJET PCR Cloning Kit (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: K1231 )
- LB agar, Miller (BD, DifcoTM, catalog number: 244520 )
- LB broth, Miller (BD, DifcoTM, catalog number: 244620 )
- Tris (hydroxymethyl) aminomethane (NACALAI TESQUE, catalog number: 35434-21 )
- Potassium chloride (Wako Pure Chemical Industries, catalog number: 163-03545 )
- Ethylenediaminetetraacetic acid (EDTA) (DOJINDO, catalog number: 345-01865 )
- Acetic acid (Wako Pure Chemical Industries, catalog number: 017-00256 )
- Ethidium bromide solution (NACALAI TESQUE, catalog number: 14631-94 )
- LB agar plate with 50 µg/ml ampicillin (see Recipes)
- Lysis buffer (see Recipes)
- 50 x TAE buffer (see Recipes)
- 1x TAE buffer (see Recipes)
Equipment
- Plant growth chamber (SANYO, model: MLR-350HT )
- Micropipettes (Eppendorf, model: Reference® 2 )
- Sterile tweezers (Electron Microscopy Sciences, DUMONT, catalog number: 0108-5-PO )
- Thermal cycler (PCR Thermal Cycler Dice® Gradient, Takara Bio, model: TP600 )
- High-speed centrifuge (TOMY DIGITAL BIOLOGY, model: MX-300 )
- Block incubator (ASTEC, model: Bl-515A )
- Spectrophotometer (GE Healthcare, model: NanoVue )
- Autoclave (TOMY DIGITAL BIOLOGY, model: SX300 )
- Agarose gel electrophoresis equipment (Cosmo Bio, model: i-MyRun.N )
- Ultraviolet transilluminator (ATTO, models: DTB-20MP , TYPE-CX )
- Heated incubator (SANYO, model: MIR-262 )
Procedure
- Design and construction of sgRNA expression vector
- Selection of target sequence
- Choose the 20 bp target sequence in the gene or genomic region of interest (Figure 1B).
- Check the protospacer-adjacent motif (PAM) sequence (5’-NGG-3’) at the 3’-end of the target sequence.
- To induce a dysfunctional mutation in the gene, the target sequence should be designed near the 5’-end or in the functional domain.
- Avoid poly-T sequences, which work as transcriptional terminators for sgRNA; ensure that the G + C content is 40-80%.
- If the target site includes the recognition site of a restriction enzyme, it will be possible to perform restriction fragment length polymorphism (RFLP) analysis for genotyping.
Note: It has been reported that truncated sgRNA could decrease off-target effects (Fu et al., 2014). We confirmed that truncated sgRNA (18 or 17 bp target sequences) works in our system; however, we have not yet evaluated their targeting efficiency.
- Check the protospacer-adjacent motif (PAM) sequence (5’-NGG-3’) at the 3’-end of the target sequence.
- Add the following adapter sequence for cloning (Figure 1B): 5’-TCTG-3’ for sense oligonucleotides and 5’-AAAC-3’ for anti-sense oligonucleotides.
- Choose the 20 bp target sequence in the gene or genomic region of interest (Figure 1B).
- Digestion of pSCgRNA vector (Figure 1C)
- Prepare the digestion mixture, as given below:

- Incubate the mixture at 37 °C for 3 h.
- Purify the BbsI-digested pSCgRNA vector using the DNA purification kit (Favorgen GEL/PCR Purification Mini Kit).
- Prepare the digestion mixture, as given below:
- Annealing of oligonucleotides (Figure 1C)
- Prepare the annealing mixture, as given below:

- Incubate at 95 °C for 5 min in a thermal cycler.
- Remove the tube from the thermal cycler and allow the mixture to cool down on the bench at 20-25 °C for 30 min.
- Prepare the annealing mixture, as given below:
- Cloning of annealed oligonucleotides into pSCgRNA vector (Figure 1C)
- Prepare the ligation mixture, as given below:

- Incubate the mixture at 16 °C for 3 h or more.
- Transform DH5α-competent cells using 2 µl ligation mixture and spread on an LB plate with 50 µg/ml ampicillin.
- Incubate the LB plate at 37 °C for 14-16 h.
- Mix a PCR reaction mixture, using the ScU6p Ins. check F primer (as the forward primer) and the antisense target sequence (as the reverse primer).

- Verify the insertion by performing a colony PCR with the following cycle:

Note: If the cloning is successful, a 950 bp band will be amplified and detected (Figure 1D). We recommend testing at least 8-16 colonies. - Grow the positive colonies in liquid LB medium with 50 µg/ml ampicillin.
- Purify the plasmids using a plasmid DNA mini prep kit (Favorgen Plasmid DNA Extraction Mini Kit).
- Confirm the inserted target sequence in the pSCgRNA vector using ScU6p Seq. check F primer using Sanger sequencing.

Figure 1. Vector construction for targeted mutagenesis in mosses using RNA-guided endonucleases. A. Schematic illustration of the Cas9 and sgRNA expression vectors; B. Example of target sequence design; C. Procedure for constructing sgRNA expression vector; D. Representative results of colony-PCR for checking the insert target sequence. M: DNA ladder markers.
- Prepare the ligation mixture, as given below:
- Selection of target sequence
- PEG-mediated protoplast transformation (Figure 2A)
- Purify the pSCgRNA containing the target sequence and pSCOE1-fcoCas9, which backbone is pTN182 vector (Sakakibara et al., 2008), using the midi prep kit (NucleoBond® Xtra Midi) and dilute it with sterile TE buffer at 1 µg/µl. Use 15 µg of each plasmid for transformation.
- Co-transform both the vectors using the PEG-mediated protoplast transformation method, as described in a previous report (Nishiyama et al., 2000; PHYSCOmanual ver. 2.0 in NIBB PHYSCObase website: http://moss.nibb.ac.jp/protocol.html).
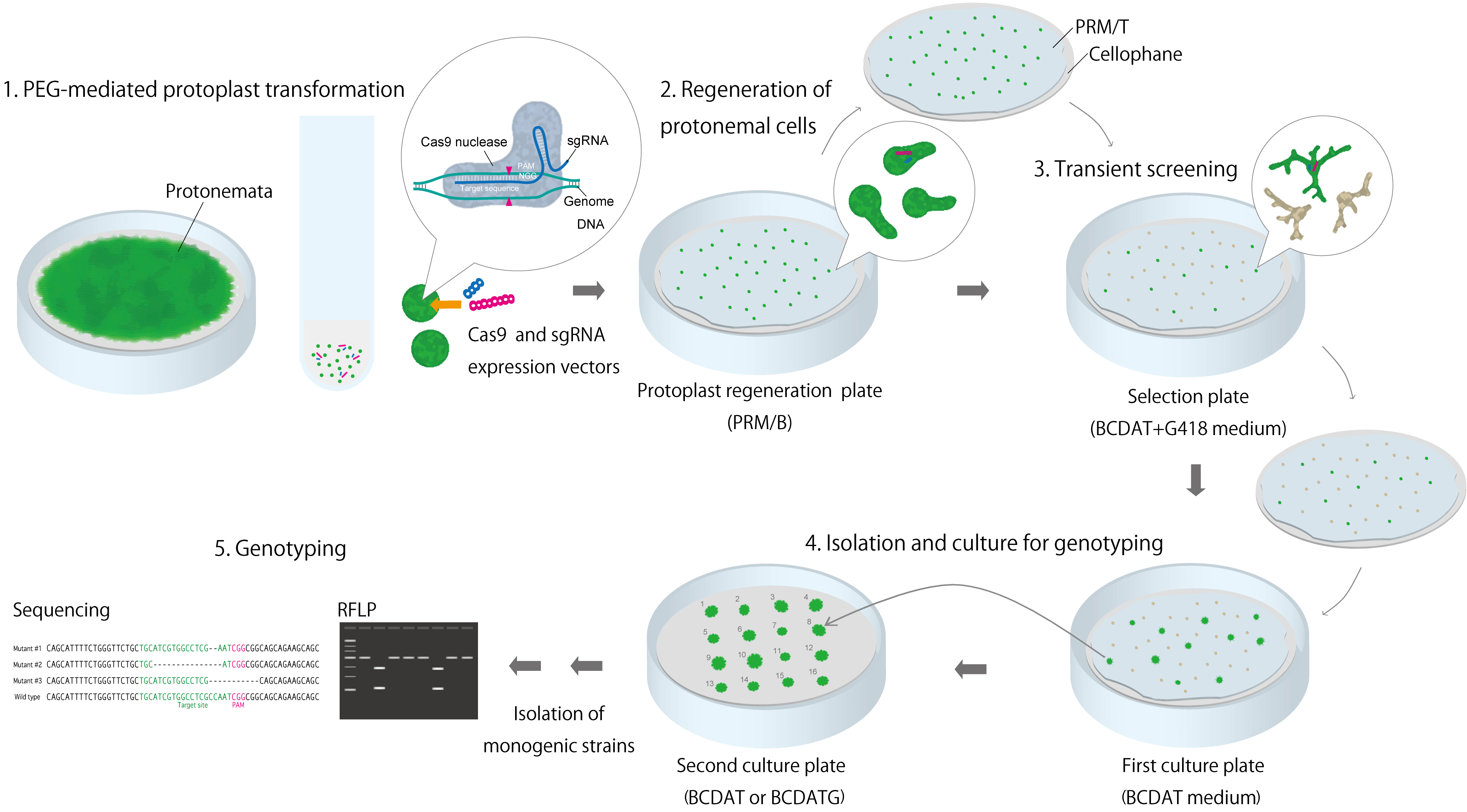
Figure 2. Overview of protocol for acquiring mutant strains using RGENs
- Purify the pSCgRNA containing the target sequence and pSCOE1-fcoCas9, which backbone is pTN182 vector (Sakakibara et al., 2008), using the midi prep kit (NucleoBond® Xtra Midi) and dilute it with sterile TE buffer at 1 µg/µl. Use 15 µg of each plasmid for transformation.
- Regeneration of protonemal cells and transient screening using antibiotic G418 (Geneticin).
- After inoculating the transformed protoplasts in PRM/T on cellophane and on a PRM/B plate, incubate the mixture at 25 °C for 7 d in a plant growth chamber (Figure 2B).
- For transient screening, transfer the regenerated protonemata from the protoplasts in PRM/T on cellophane to new BCDAT plates with 20 µg/ml G418 (Figure 2C).
- Culture the plates in a plant growth chamber for 5 d (Figures 2C and 3).
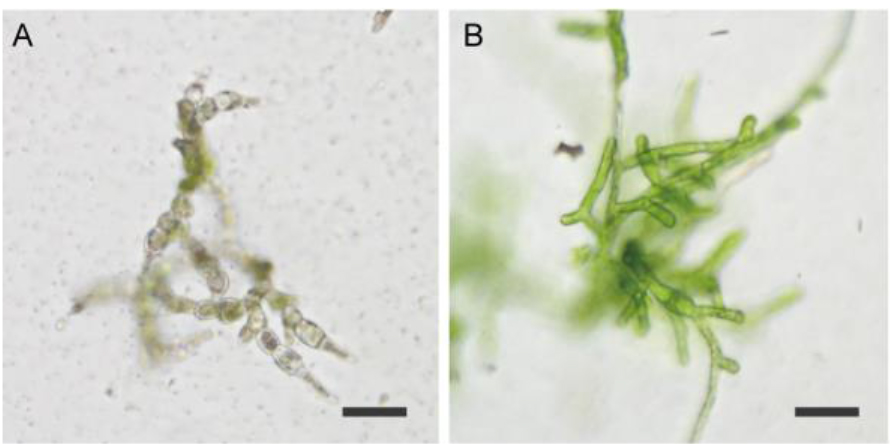
Figure 3. Example of protonemata after transient screening (PpFtsZ2-1 targeted). A. Representative example of dead protonemata; B. Representative example of surviving protonemata. Strain also shows the phenotype of PpFtsZ2-1 mutation. Scale bars = 50 µm. - Transfer the surviving protonemata in PRM/T on cellophane to new BCDAT plates and culture again until small colonies have formed (Figure 2D).
- Using tweezers, choose the protonemata at the edge of the colonies and transfer them to new, numbered BCDAT or BCDAT + 0.5% glucose plates (Figure 2D).
- For genotyping, culture the protonemata until the colonies become 5-10 mm in diameter.
Note: There is a possibility that the acquired strain is a mosaic plant, which is a mixture of strains with different mutations or wild type. If so, we can isolate the monogenic strain from the protoplast, single protonemal branch, or leaf of the gametophore.
- After inoculating the transformed protoplasts in PRM/T on cellophane and on a PRM/B plate, incubate the mixture at 25 °C for 7 d in a plant growth chamber (Figure 2B).
- Genotyping
- Green PCR
- Transfer a pinch of fresh protonemata to PCR tubes (Figure 4A) and grind using the tip of the 200 µl pipette chip into 50 µl lysis buffer (see Recipes) (Figure 4B).

Figure 4. Preparation of the template for Green PCR. A. A pinch of fresh protonemata in the lysis buffer; B. Protonemata crushed by the tip of the 200 µl chip in the lysis buffer. - Incubate at 95 °C for 10 min.
- Spin down the debris at 3,500 x g for 10 sec and use the supernatant as the PCR template.
- Design the primer set using free online tool Primer 3 (http://primer3.ut.ee/) for amplifying the genomic DNA, including the target site for sequencing or for other genotyping such as RFLP.
- PreparePCR reaction mixture, as given below:

- Amplify the PCR product, including the target site of the genome, with the cycle given below:

- Transfer a pinch of fresh protonemata to PCR tubes (Figure 4A) and grind using the tip of the 200 µl pipette chip into 50 µl lysis buffer (see Recipes) (Figure 4B).
- Restriction fragment length polymorphism (RFLP) analysis
- If the target site includes the recognition site of a restriction enzyme, it will be possible to observe the targeted mutagenesis by RFLP.
- After confirming the PCR products by agarose gel electrophoresis, digest the PCR products using the corresponding restriction enzymes.
- After sufficient digestion, analyze the enzyme-treated and untreated PCR products by agarose gel electrophoresis. It is possible that restriction enzyme-tolerant PCR products have mutations.
Note: If a mixture of PCR products derived from wild type and mutant strains is used, the targeted mutagenesis can be checked using the T7 endonuclease I or cel1 assay, which can recognize and cleave non-perfectly matched DNA. As high throughput and comprehensive genotyping method, high resolution melting analysis or next-generation sequencing is also applicable.
- If the target site includes the recognition site of a restriction enzyme, it will be possible to observe the targeted mutagenesis by RFLP.
- Sequencing of target sites in the genome
- After confirming the PCR product by agarose gel electrophoresis, clone the PCR products into the sequencing vector using the Zero Blunt PCR Cloning Kit or the CloneJET PCR Cloning Kit.
- After colony PCR and mini prep, check the DNA sequence of the inserted PCR product of several clones by Sanger sequencing.
Note: If the PCR product is present as a single band in monogenic plant, it will be possible to check by direct sequencing.
- After confirming the PCR product by agarose gel electrophoresis, clone the PCR products into the sequencing vector using the Zero Blunt PCR Cloning Kit or the CloneJET PCR Cloning Kit.
- Green PCR
Data analysis
According to our evaluation based on the phenotypic change caused by the mutation of a target gene, the target mutagenesis efficiency in P. patens was approximately 45% to 68% (Nomura et al., 2016). Since efficiency is dependent on the target sequence, we recommend using the ‘focas’ website (http://focas.ayanel.com/; Doench et al., 2014; Xiao et al., 2014; Osakabe et al., 2016) for designing and predicting the on- and off-target efficiency of the target genome. In this case, the target sequence with an on-target score of 0.6 or higher and a low possibility of off-target effect is desirable.
Notes
- It is possible to introduce long deletion mutations (~3 kb) through this method by using two gRNAs designed for distant sites on the genome (Nomura et al., 2016).
- In this method, the vector DNA is usually not integrated into the moss genome (Nomura et al., 2016).
- There is the possibility of the formation of polyploidy during the PEG-mediated protoplast transformation. DNA content in acquired strains should be assessed using a flow cytometer.
- If this protocol is applied to other species, it may be necessary to modify some experimental conditions such as the transformation (e.g., PEG treatment time) and screening method (e.g., antibiotic concentration and duration of selection).
Recipes
- LB agar plate with 50 µg/ml ampicillin
LB agar, Miller 40 g
Fill up to 1 L with Milli-Q water and autoclave (121 °C, 15 min)
Add ampicillin to a final concentration of 50 µg/ml - Lysis buffer
100 mM Tris-HCl (pH 9.5)
1 M KCl
10 mM EDTA - 50 x TAE buffer
2 M Tris
1 M acetic acid
0.5 M EDTA (pH = 8.0) - 1 x TAE buffer
20 ml 50 x TAE buffer
980 ml Milli-Q water
Acknowledgments
This protocol was adapted from a published paper (Nomura et al., 2016). This work was supported by the Japan Society for the Promotion of Science, Grant-in-Aid for Young Scientists (B) (grant number 15K18824), and Grants-in-Aid for Scientific Research (C) (grant number 15K06905).
References
- Doench, J. G., Hartenian, E., Graham, D. B., Tothova, Z., Hegde, M., Smith, I., Sullender, M., Ebert, B. L., Xavier, R. J. and Root, D. E. (2014). Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol 32(12): 1262-1267.
- Fu, Y., Sander, J. D., Reyon, D., Cascio, V. M. and Joung, J. K. (2014). Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32(3): 279-284.
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A. and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096): 816-821.
- Nishiyama, T., Hiwatashi, Y., Sakakibara, I., Kato, M. and Hasebe, M. (2000). Tagged mutagenesis and gene-trap in the moss, Physcomitrella patens by shuttle mutagenesis. DNA Res 7(1): 9-17.
- Nomura, T., Sakurai, T., Osakabe, Y., Osakabe, K. and Sakakibara, H. (2016). Efficient and heritable targeted mutagenesis in mosses using the CRISPR/Cas9 system. Plant Cell Physiol 57(12): 2600-2610.
- Osakabe, Y., Watanabe, T., Sugano, S. S., Ueta, R., Ishihara, R., Shinozaki, K. and Osakabe, K. (2016). Optimization of CRISPR/Cas9 genome editing to modify abiotic stress responses in plants. Sci Rep 6: 26685.
- Sakakibara, K., Nishiyama, T., Deguchi, H. and Hasebe, M. (2008). Class 1 KNOX genes are not involved in shoot development in the moss Physcomitrella patens but do function in sporophyte development. Evol Dev 10:555-566.
- Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M. and Rozen, S. G. (2012). Primer3—new capabilities and interfaces. Nucleic Acids Res 40(15): e115-e115.
- Xiao, A., Cheng, Z., Kong, L., Zhu, Z., Lin, S., Gao, G. and Zhang, B. (2014). CasOT: a genome-wide Cas9/gRNA off-target searching tool. Bioinformatics 30:1180-1182.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Nomura, T. and Sakakibara, H. (2017). Targeted Mutagenesis Using RNA-guided Endonucleases in Mosses. Bio-protocol 7(12): e2359. DOI: 10.21769/BioProtoc.2359.
Category
Plant Science > Plant molecular biology > DNA > Mutagenesis
Molecular Biology > DNA > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.