- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Single Genome Sequencing of Expressed and Proviral HIV-1 Envelope Glycoprotein 120 (gp120) and nef Genes

Published: Vol 7, Iss 12, Jun 20, 2017 DOI: 10.21769/BioProtoc.2334 Views: 10060
Reviewed by: Chao JiangYi ZhangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The current study provides detailed protocols utilized to amplify the complete HIV-1 gp120 and nef genes from single copies of expressed or integrated HIV present in fresh-frozen autopsy tissues of patients who died while on combined antiretroviral therapy (cART) with no detectable plasma viral load (pVL) at death (Lamers et al., 2016a and 2016b; Rose et al., 2016). This method optimizes protocols from previous publications (Palmer et al., 2005; Norström et al., 2012; Lamers et al., 2015; 2016a and 2016b; Rife et al., 2016) to produce single distinct PCR products that can be directly sequenced and includes several cost-saving and time-efficient modifications.
Keywords: HIV-1Background
Over thirty years ago, HIV infection and its clinical manifestation, Acquired Immunodeficiency Syndrome (AIDS), emerged as a worldwide epidemic. Since then, significant understanding of HIV pathogenesis has occurred and the development of drug treatments now significantly extend patients’ lives. Current cART regimens encompass a variety of drugs that inhibit viral replication in several ways, which allows for the almost complete suppression of viral particles found in the blood and recovery of a healthy CD4+ T-cell population (CD4+) (Autran et al., 1997). However, the persistence of very low levels of HIV in plasma of cART treated patients, even those treated for decades, suggests the presence of a cell based ‘viral reservoir’. Viral reservoirs contain infected cells that do not release infectious virus (i.e., are latently infected), but can do so following activation, which may occur under a variety of conditions (Chun et al., 1995 and 1997). HIV latency is primarily attributed to proviral HIV DNA in resting memory CD4+ T cells (Anderson et al., 2011; Ho et al., 2013), although recent reviews highlight a breadth of research into other potential reservoirs (Abbas et al., 2015; Kandathil et al., 2016; Rothenberger et al., 2016; Sacha and Ndhlovu, 2016). The resting memory CD4+ T cells can live for long periods of time, contribute to low-level persistent viremia during cART and viral rebound after treatment interruption, and produce viral variants with escape mutations (Chun et al., 1997; Finzi et al., 1997). Methods to determine the effectiveness of antiretroviral therapy and latency-reversing agents by measuring the circulating resting memory CD4+ T cells have been developed and evaluated (Ericksson et al., 2013; Crooks et al., 2015). However, it is pertinent to consider that less than 2% of the total body lymphocyte population resides in peripheral blood (Svincher et al., 2014), making the evaluation of HIV persistence of tissue-resident lymphocyte populations in anatomical reservoirs critically important.
The use of single genome sequencing or SGS (also known as single genome amplification or SGA) has become the routine way to generate sequences for examination of HIV intrahost evolution (Kearney et al., 2014; Lamers et al., 2016; Rose et al., 2016), compartmentalization (Sturdevant et al., 2012; Evering et al., 2014), phyloanatomy (Salemi and Rife, 2016), persistence (Josephsson et al., 2013; Buzon et al., 2014; Boritz et al., 2016), and rebound dynamics (Kearney et al., 2015; Bednar et al., 2016). In contrast to bulk PCR methods wherein many targets are amplified together in the same tube, SGS uses end-point dilution to amplify from only one template. While some studies have demonstrated that bulk PCR and SGS produce sequences that are similar by certain metrics and the techniques can be used interchangeably (Jordan et al., 2010; Etemad et al., 2015), some analyses can only yield accurate results with sequences generated from SGS. These include identifying identical HIV sequences that may arise from clonally-expanding cells rather than PCR resampling (Wagner et al., 2013; Simonetti et al., 2016), determining proportions of viral variants in a sample through sequencing (Iyer et al., 2015), estimating evolutionary rate from point-mutations that occur only from viral reverse-transcriptase rather than PCR Taq errors (Novitsky et al., 2013), and evaluating recombination rates in vivo without including PCR-mediated recombination (Brown et al., 2011; Sanborn et al., 2015).
We used SGS to generate linked gp120 envelope and nef gene sequences from single starting templates to assess viral expression, compartmentalization and evolution in RNA and DNA extracted from a collection of fresh frozen tissues obtained from HIV-infected patients on cART who died with no detectable viral load in their plasma or cerebral spinal fluid at the time of death (Lamers et al., 2016a and 2016b; Rose et al., 2016). Our data demonstrated that a privileged environment exists in some tissues of these patients wherein expression of HIV continues; however, in other tissues, only unexpressed proviral DNA copies were identified. The inferred evolutionary rate of the tissue-based HIV sequences was not significantly different than previously reported rates of replicating virus in cART-negative subjects, suggesting on-going evolution.
Materials and Reagents
- RNA and DNA extraction
- Pipette tips
- TissueRuptor disposable probes (QIAGEN, catalog number: 990890 )
- Fresh frozen tissue sections (30-50 ng)
- ELIMINaseTM Decontaminant (Fisher Scientific, catalog number: 04-355-32 )
- AllPrep DNA/RNA Mini Kit (QIAGEN, catalog number: 80204 )
- RNeasy MinElute Cleanup Kit (QIAGEN, catalog number: 74204 )
- Qubit 2.0 fluorometer (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32857 )
- Ethyl alcohol pure (200 Proof molecular biology grade) (Sigma-Aldrich, catalog number: E7023 )
- Qubit® dsDNA HS Assay Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32854 )
- Qubit® RNA HS Assay Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32852 )
- Pipette tips
- cDNA synthesis
- 0.2 ml PCR 8-tube FLEX-FREE strip, attached clear flat caps, natural (USA Scientific, catalog number: 1402-4700 )
- SuperScript® III First-Strand Synthesis System (Thermo Fisher Scientific, InvitrogenTM, catalog number: 18080051 ). The SuperScript® III First-Strand Synthesis System is supplied with the following:
- Oligo(dT)20 (50 µM), 50 µl
- Random hexamers (50 ng/µl), 250 µl
- 10x RT buffer, 1 ml
- 0.1 M DTT, 250 µl
- 25 mM magnesium chloride (MgCl2), 500 µl
- 10 mM dNTP mix, 250 µl
- SuperScript® III RT (200 U/µl), 50 µl
- RNase-OUTTM (40 U/µl), 100 µl
- E. coli RNase H (2 U/µl), 50 µl
- DEPC-treated water, 1.2 ml
- Total HeLa RNA (10 ng/µl), 20 µl
- Sense Control Primer (10 µM), 25 µl
- Antisense Control Primer (10 µM), 25 µl
- Oligo(dT)20 (50 µM), 50 µl
- 0.2 ml PCR 8-tube FLEX-FREE strip, attached clear flat caps, natural (USA Scientific, catalog number: 1402-4700 )
- Single genome sequencing of gp120 and nef
- 24 PCR wells
- Pipette tips
- TempPlate semi-skirted polypropylene 0.2 ml 96-well PCR plate (USA Scientific, catalog number: 1402-9220 )
- Posi-Click 1.7 ml microcentrifuge tube, 1.7 ml natural color (Denville Scientific, catalog number: C2170 )
- Molecular biology grade sterile purified water (RNase, DNase, proteinase free)
- EB buffer (QIAGEN, catalog number: 19086 )
- Platinum® Blue PCR SuperMix (Thermo Fisher Scientific, InvitrogenTM, catalog number: 12580023 )
- Agarose (Fisher Scientific, catalog number: BP160-500 )
- Ethidium bromide (Fisher Scientific, catalog number: BP102-1 )
- Tris-base (Sigma-Aldrich, catalog number: T1378 )
- Acetic acid, glacial (Fisher Scientific, catalog number: A38-212 )
- Ethylenediaminetetraacetic acid, EDTA, 0.5 M solution/pH 8.0 (Fisher Scientific, catalog number: BP2482-500 )
- Milli-Q quality water (RNase, DNase free water [dH2O])
- Primers listed in Table 1
Table 1. Primers
- 50x TAE stock solution (see Recipes)
- 1x TAE buffer(see Recipes)
- 24 PCR wells
Equipment
- TissueRupter rotor-stator homogenizer (QIAGEN, model: TissueRupter, catalog number: 9001271 )
- Matrix multichannel electronic pipette (Range: 2-125 µl; 12-channel) (Fisher Scientific, catalog number: 14-387-117 )*
- Matrix multichannel electronic pipette (Range: 1-30 µl; 12-channel) (Thermo Fisher Scientific, catalog number: 14-387-137 )*
- Matrix multichannel electronic pipette (Range: 2-125 µl; 12-channel) (Thermo Fisher Scientific, catalog number: 14-387-138 )*
- Eppendorf RepeaterTM stream electronic pipette (Eppendorf, catalog number: 4987000118 )
- Eppendorf ResearchTM Plus adjustable-volume pipettes: 0.1-2.5 µl, 2-20 µl, 20-200 µl, 100-1,000 µl (Eppendorf, catalog number: 022575442 )
- Tape pads (QIAGEN, catalog number: 19570 )
- Sub-CellTM Model 192 electrophoresis system (Bio-Rad Laboratories, model: Model 192, catalog number: 1704507 )
- 51-Well comb (Bio-Rad Laboratories, catalog number: 1704529 )
- Comb holder (Bio-Rad Laboratories, catalog number: 1704525 )
- UV-Transparent gel tray (Bio-Rad Laboratories, catalog number: 1704524 )
- Model 192 gel caster (Bio-Rad Laboratories, model: Model 192, catalog number: 1704517 )
- Centrifuge 5424, non-refrigerated, with Rotor FA-45-24-11, keypad, 230 V/50 -60 Hz (Eppendorf, model: 5424 , catalog number: 5424000010)
- IsotempTM Digital Dry Bath incubator (Fisher Scientific, catalog number: 11-718-2Q )*
- T100TM Thermal cycler (Bio-Rad Laboratories, model: T100TM, catalog number: 1861096 )
- DNA oligonucleotides were obtained from Invitrogen
- Applied Biosystems 3730xl DNA analyzer (Thermo Fisher Scientific, Applied BiosystemsTM, model: 3730xl DNA Analyser , catalog number: 3730XL)
*Note: These products have been discontinued.
Software
- Geneious R7 software package (Biomatters http://www.geneious.com)
- MEGA5
Procedure
- RNA and DNA extraction
- Thoroughly clean work surfaces and equipment before and after use with ELIMINase Decontaminant.
Note: RNA and DNA extractions, cDNA synthesis and first round PCR set-up should be performed using filtered pipette tips and must be conducted in a restricted-access amplicon-free room with separate air-handling and laboratory equipment where no amplified PCR products or recombinant cloned plasmids are allowed. If no such room is available, conduct steps before amplification in a cell biology-grade clean hood equipped with separate air-handling mechanisms. - Total RNA and genomic DNA are isolated separately and simultaneously from each tissue section (30-50 ng) using the AllPrep DNA/RNA Mini Kit following manufacturer’s guidelines. Two final 50 μl elutions using RNase-free water are performed during the last step of RNA isolation, totaling a final volume of 100 μl.
- Tissues are homogenized just prior to extraction using a TissueRupter rotor-stator homogenizer with a fresh sterile disposable probe for each sample.
- The 100 μl final volume of RNA is concentrated using RNeasy MinElute Cleanup Kit according to manufacturer’s instructions. A single final elution of 20 μl RNase-free water is used.
- Quantification of the resulting RNA and DNA is performed to determine the success of the extraction protocol and the concentration, utilizing the Qubit 2.0 fluorometer and either the Qubit RNA HS Assay Kit or Qubit dsDNA HS Assay Kit where appropriate. Failure to detect DNA or RNA, or a yield of less than 1 ng/μl for either RNA or DNA, indicates a failed extraction and the extraction should be repeated until more than 1 ng/μl of RNA and DNA is detected.
- Thoroughly clean work surfaces and equipment before and after use with ELIMINase Decontaminant.
- cDNA synthesis
- cDNA is created immediately from the RNA of each sample using the SuperScript® III First-Strand Synthesis System using the provided oligo(dT)20 primer according to manufacturer’s recommendations with slight modifications, detailed below, to increase product length.
- In two identical reactions for each sample, 8 μl RNA is incubated at 65 °C for 5 min with deoxynucleoside triphosphates (0.5 mM [each]) and 5 µM oligo (dT)20, then cooled quickly to 4 °C.
Note: Use thermocycler for accurate temperatures and hold times. cDNA synthesis reactions are conducted in 0.2 ml PCR 8-tube FLEX-FREE strips. - First-strand cDNA synthesis will continue in a 20 µl reaction volume containing 1x reverse transcription buffer (10 mM Tris-HCl [pH 8.4], 25 mM KCl), 5 mM MgCl2, 10 mM ditiothreitol, 2 U/µl of RNase-OUTTM (RNase inhibitor), and 10 U/µl SuperScript® III RT. The reaction is heated to 45 °C for 90 min, and then 85 °C for 5 min.
- The reaction is then cooled to 37 °C and 0.1 U/µl of E. coli RNase H is added, followed by a 20-min incubation.
- The two reactions for each sample are combined with gentle pipette mixing to avoid shearing the cDNA. cDNA is stored at -20 °C until needed.

- cDNA is created immediately from the RNA of each sample using the SuperScript® III First-Strand Synthesis System using the provided oligo(dT)20 primer according to manufacturer’s recommendations with slight modifications, detailed below, to increase product length.
- Single genome sequencing of gp120 and nef
- cDNA and genomic DNA (gDNA) dilutions using EB buffer are usually performed to achieve 30% or less of positive nested PCR reactions, which indicates the positive reactions will have a greater than 80% chance of one starting template.
Notes: - For patients on cART, it is practical to start with 1:3 and 1:9 dilutions of cDNA and gDNA, with 24 PCR wells for each dilution. For patients not on cART, higher dilutions can be used.
- Stock and dilutions must be kept on ice after thawing and mixing, and frozen at -20 °C when not in use. Pipette mix or flick mix samples and dilutions, do not vortex to mix.
- Serial dilutions and first round PCR setup must be done in the amplicon-free room and always use filtered pipette tips.
- Two rounds of PCR are required to generate enough product for visualization, quantification and sequencing when starting with only a single template.
- During the first round PCR, 1 µl of diluted cDNA or genomic DNA is amplified in 20 µl reactions containing 1x Platinum® Blue PCR SuperMix and 0.05 µM of each primer: BEF1, 5’-TAATAGCAATAGTTGTGTGG-3’ and BNR1, 5’-AGCTCCCAGGCTCAGATCT-3’ (6,111-6,130 and 9,558-9,576 bp of HIV-1 HXB2 respectively).
- The first round primers are at 0.05 µM concentration in the reaction volume to eliminate unused excess first round primer carryover into the second round PCR. Excess first round primers in the second round PCR produces non-specific PCR products and reduces the amount of the desired product. See Figure 1 for an example of the non-specific PCR products generated by first round PCR primer carryover.
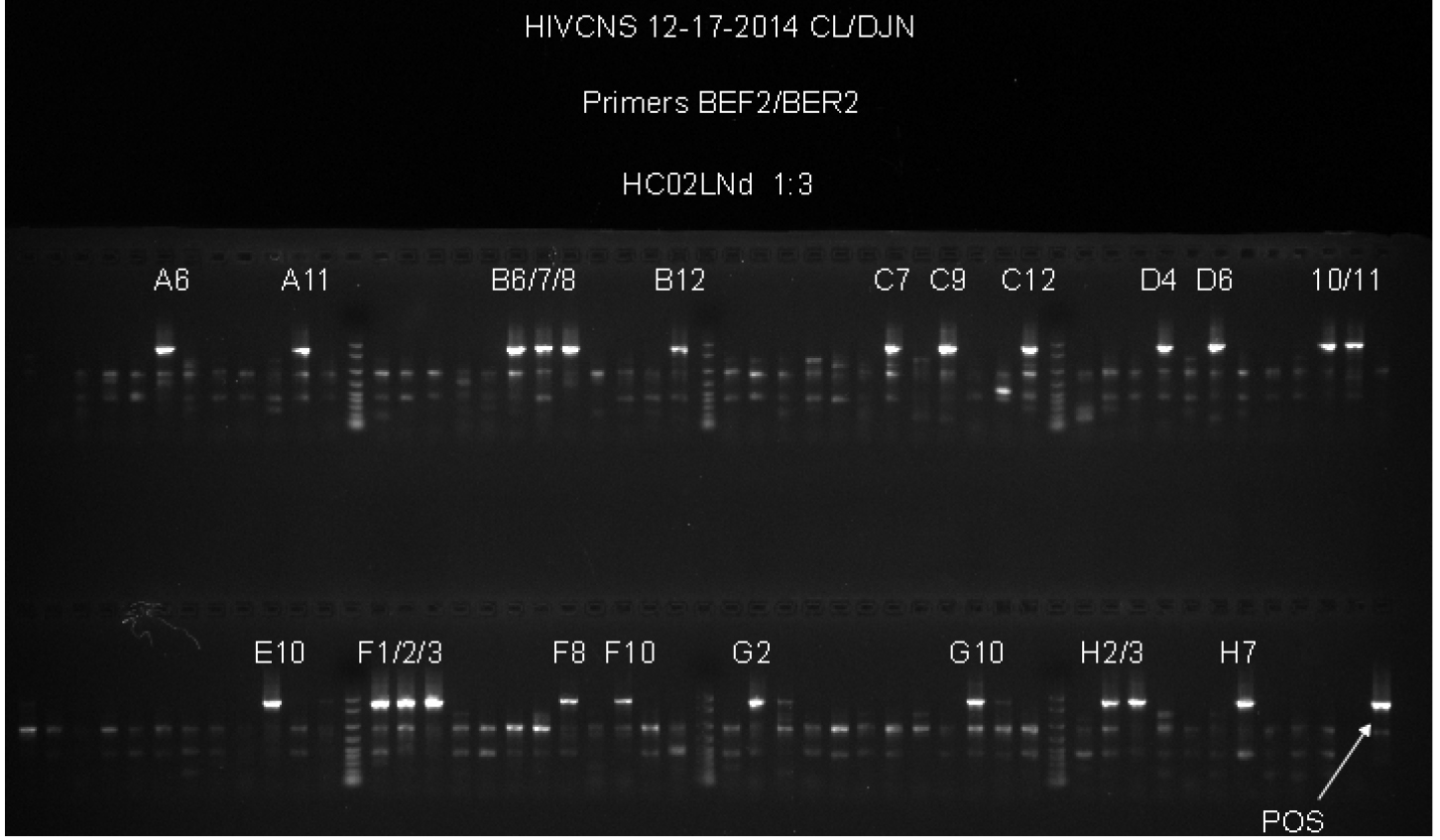
Figure 1. Example of non-specific primer binding to genomic DNA. This gel provides an example of experiments where the concentration of the first round PCR primers was 0.2 µM in the first round PCR. First round primers at this concentration resulted in non-specific product formation in the second round PCR, as seen by the fainter bands found in most wells, whether or not those wells have a bright band that corresponds to the size of the positive control. When the first round PCR primers were used at 0.05 µM, these secondary products are no longer visible while the positive PCR products of the correct size are still visible, resulting in easier to interpret results and direct sequencing of the second round products without further gel purification to isolate a single band.
- The first round primers are at 0.05 µM concentration in the reaction volume to eliminate unused excess first round primer carryover into the second round PCR. Excess first round primers in the second round PCR produces non-specific PCR products and reduces the amount of the desired product. See Figure 1 for an example of the non-specific PCR products generated by first round PCR primer carryover.
- First round PCR cycling parameters–an initial denaturation 94 °C for 3 min, then 40 cycles of 94 °C for 30 sec, 56 °C for 30 sec,72 °C for 4 min, followed by a final extension of 72 °C for 10 min.

Notes: - PCRs are conducted in a 96-well format using TempPlate semi-skirted polypropylene 0.2 ml 96-well PCR plates and Tape pads. Large batches of PCR plates containing premixed SuperMix and primers are created and frozen for future use to reduce inter-experiment variability.
- Positive PCR controls should be selected carefully and diluted enough to produce only a single band after nested PCR is complete–therefore a band will not be visible after the first round PCR. Very concentrated positive controls can easily contaminate the PCR plates and diluted to a workable level.
- Using automated pipettes reduces the possibility of error and cross-contamination. We use Matrix multichannel electronic pipette (Range: 1-30 µl; 12-channel) and Eppendorf Repeater stream electronic pipette.
- The amount of primer in first round PCR is reduced significantly to reduce non-specific binding and primer carry-over during second round PCR. Extension times and cycle number are increased to generate an increased number of complete products.
- Second round gp120 PCR consists of 2 µl of the first round PCR product added to a 20 µl second round reaction consisting of 1x Platinum® Blue PCR SuperMix and 0.2 µM of each primer: BEF2, 5’-CAATAGTTGTGTGGTCCATAG-3’ and BER2, 5’-CAACAGATGCTGTTGCGC-3’ (6,117-6,137 bp and 7,905-7,922 bp of HIV-1 HXB2 respectively)
- Second round gp120 PCR cycling parameters–an initial denaturation 94 °C for 3 min, then 40 cycles of 94 °C for 30 sec, 56 °C for 30 sec, 72 °C for 3 min, followed by a final extension of 72 °C for 10 min.

- Second round gp120 PCR products are visualized on 1% agarose gels stained with ethidium bromide run at 150 V for 30 min in 1x TAE buffer.
Notes:- This second round PCR generates a 1.8 Kb product when positive, containing a complete gp120 sequence. Products from positive wells are sent sequencing with BEF2 and BER2 primers. This protocol produces single specific PCR products that can be directly sequenced, and do not require PCR purification. See Figure 2 for an example of successful gp120 second round PCR with two different patient samples and a dilution series for DNA from another tissue.
- We use Platinum® Blue PCR SuperMix to direct load second round products on agarose gels rather than mixing loading dye in each reaction. We use Matrix multichannel electronic pipette (Range: 2-125 µl; 12-channel) to automated loading on a Sub-CellTM Model 192 electrophoresis system.
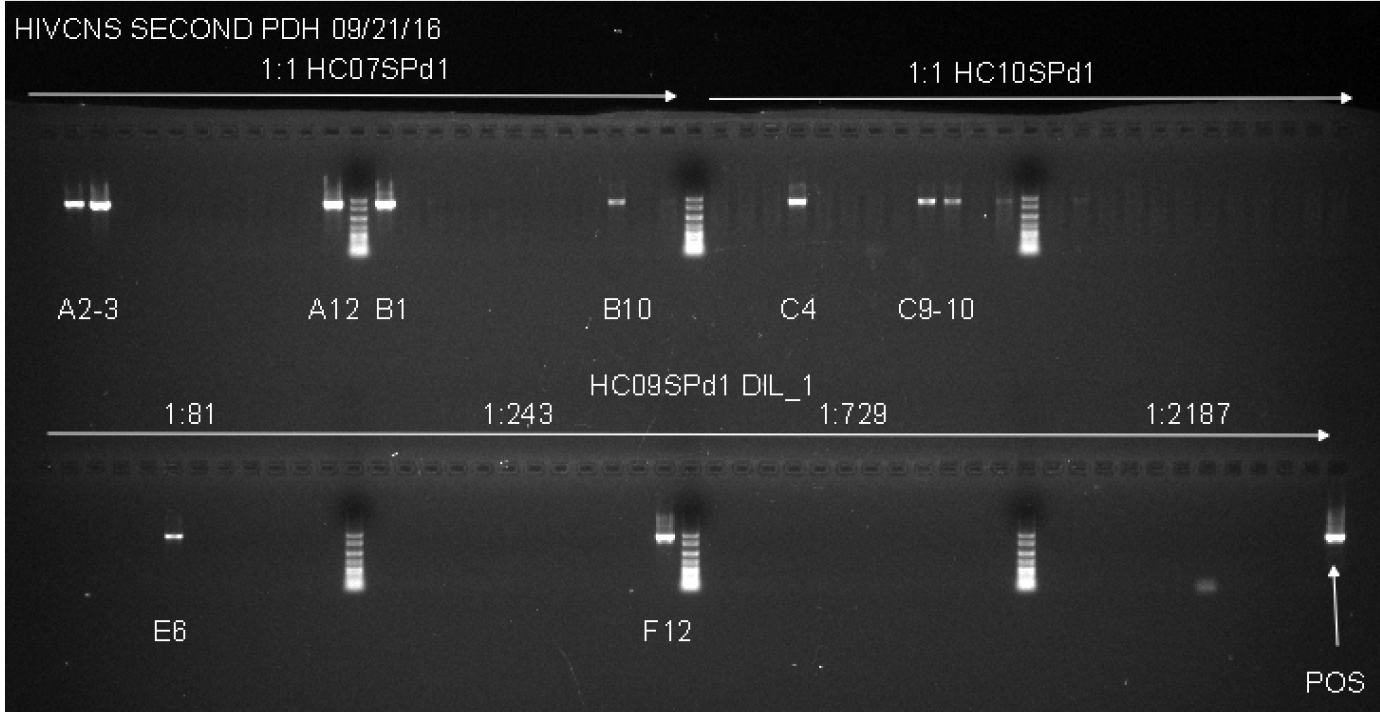
Figure 2. Example of Second Round gp120 PCR plate agarose gel image. The two samples used on the top row of the gel are undiluted genomic DNA from spleen tissue of two patients. Both samples have a total number of positive wells that equals less than 30%, indicating that the positive wells are most likely the result of nested PCR amplification of gp120 from a single integrated proviral genome in the DNA present in that well. The bottom row (HC09SPd1 DIL_1) provides an example of serial dilution testing to assess the correct SGS dilution. Four dilutions are tested here, and while all four dilutions are high enough to generate the amplification of a single integrated provirus in a positive well, all four are too high of a dilution to get many positive reactions resulting in wasted reagents. The ideal situation would be to find a dilution where 20-30% of the wells are positive, so lower dilutions must be tested to find an optimal dilution. The negative control, while not labeled on the gel, is in well A1, and the positive control (labeled POS) is in well H12. The negative control has 1 µl of the water used for dilution of the DNA, and the positive control is diluted genomic DNA from a patient who was not on cART that was PCR positive in previous experiments. - This second round PCR generates a 1.8 Kb product when positive, containing a complete gp120 sequence. Products from positive wells are sent sequencing with BEF2 and BER2 primers. This protocol produces single specific PCR products that can be directly sequenced, and do not require PCR purification. See Figure 2 for an example of successful gp120 second round PCR with two different patient samples and a dilution series for DNA from another tissue.
- Subsequently, the first round reactions that corresponded to positive second round gp120 PCRs were used to amplify the nef gene sequence; second round nef PCR consisted of 2 μl of the first round PCR added to a 20 μl second round reaction consisting of 1x Platinum® Blue PCR SuperMix and 0.2 µM of each primer: BNF1, 5’-CTGGCTGTGGAAAGATACCT-3’ and BNR2, 5’-ATCTGAGGGCTCGCCACT-3’ (7,965-7,984 bp and 9,488-9,505 of HIV-1 HXB2 respectively).
- Nef cycling parameters–an initial denaturation 94 °C for 3 min, followed by 40 cycles of 94 °C for 30 sec, 58 °C for 30 sec, 72 °C for 2 min, followed by a final extension at 72 °C for 10 min.

- Second round nef PCR products were visualized on 1% agarose gels stained with ethidium bromide run at 150 V for 30 min in 1x TAE buffer.
Note: Reactions containing single 1.5 Kb products were considered positive and selected for sequencing with BNF1 and BNR2. This protocol produces single specific PCR products that can be directly sequenced, and do not require PCR purification.
- cDNA and genomic DNA (gDNA) dilutions using EB buffer are usually performed to achieve 30% or less of positive nested PCR reactions, which indicates the positive reactions will have a greater than 80% chance of one starting template.
Data analysis
Sequencing was performed on an Applied Biosystems 3730xl DNA Analyzer at the University of Florida Interdisciplinary Center for Biotechnology Research (UF ICBR) using with BEF2/BER2 primers for gp120 and BNF1/BNR2 primers for nef sequencing. Forward and reverse chromatograms for each PCR product sequenced were assembled with the Geneious R7 software package (Biomatters http://www.geneious.com) and optimized by hand where possible to resolve ambiguous sequencing calls assigned by the sequencer or spurious gaps from the assembly algorithm. Forward and reverse chromatograms that did not assemble using the Geneious automated assembly algorithm with default settings were discarded, usually these were sequences of very poor quality (too many overlapping peaks for accurate base calling by the sequencer) or of very short length (less than 600 base pairs). For assemblies where multiple chromatogram peaks were found in two or more base pairs, indicating either multiple starting templates or multiple PCR errors in the initial amplification of the starting template, these sequences were removed from further analysis. A consensus sequence was extracted from each optimized assembly using the Geneious software package. Consensus sequences were aligned using ClustalW (Thompson et al., 1997) in MEGA5 (Tamura et al., 2011) with further optimization performed by hand to remove spurious gaps created by the alignment algorithm. The final env and nef alignments spanned from positions 6,213-7,823 and positions 8,797-9,411 relative to the HXB2 genome, respectively. Hypervariable regions in env (V1, V2 and V4 domains) were excluded due to a large number of naturally occurring insertions and deletions that are typically problematic to align and may bias phylogenetic analysis. A preliminary maximum-likelihood phylogeny for each gene was estimated using PhyML (http://www.atgc-montpellier.fr/phyml/) and sequences from all participants to ensure no cross-contamination of patients occurred. Sequences were tested for the presence of hyper-mutations using the HYPERMUTE tool (http://www.lanl.gov); sequences with a P-value of < 0.01 were removed from the alignments. Example sequences generated with this protocol have been submitted to GenBank (Accession numbers KU708874-KU709831).
Notes
When considering the results of SGS experiments, it is important to keep in mind several ideas:
- Primer binding efficiency might vary by patient, subtype, or viral gene targeted based on variations of the viral genome. Screening each patient with multiple sets of primers specific for the subtype of the patient and finding concordant results will increase confidence in the sequencing results. Tissues found to be negative for the SGS protocol for gp120-nef presented here should also be assessed with primers in more conserved regions of the HIV genome like gag (Norström et al., 2012) or pol (Palmer et al., 1999; Shafer et al., 2000) to confirm the absence of virus. Using a program like QUALITY (Rodrigo et al., 1997) to estimate copy number based on SGS dilutions (Rife et al., 2016) with SGS results from different primer sets can also provide data on the binding efficiency of each set. In addition, alternative gp120/nef primers (Lamers et al., 2016b) can be used to confirm that some variants are not missed due to primer binding efficiency of the primers presented here. Sequences generated from these alternative primers can be included in phylogenic analysis to evaluate the efficiency of the original primers at capturing the landscape of viral diversity in the tissue. Real-time or quantitative PCR can also be used to evaluate positive or negative SGS results (Lamers et al., 2016a).
- Tissue type can affect the DNA and RNA isolation. The Qiagen Allprep kit has some detailed instructions on altering methods to boost isolation efficiency for different tissue types. Alternative kits or protocols should be considered for tissues that consistently result in low yields. Tissue preservation will also affect isolation results and great care in handling tissues should be exercised to prevent premature thawing. Separate isolations conducted on multiple tissue sections will increase confidence in SGS results. Tissues from the same patient should be processed separately where possible, as cross-contamination of samples from the same patient will not be as easily recognized as mixing between patients during initial phylogenetic analysis of all sequences generated.
Recipes
- 50x TAE stock solution
To prepare 1 L of 50x TAE dissolve following components:
in 600 ml of deionized water:
242 g Tris base (FW = 121)
57.1 ml glacial acetic acid
100 ml 0.5 M EDTA (pH 8.0) - 1x TAE buffer
40 mM Tris (pH 7.6)
20 mM acetic acid
1 mM EDTA
Dilute 1:50 for 1x TAE buffer for gel electrophoresis
Acknowledgments
Funding for development of this HIV-1 sequencing protocol was provided by the National Institutes of Health (NIH) through the R01 MH100984 grant, and is based on methods developed on a previous NIH grant, R01 NS063897. We would like to thank the National Neurological AIDS Bank (NNAB, UM1 CA181255) and the AIDS and Cancer Specimen Resource (ACSR, UM1 CA181255) to providing the tissues used to develop the protocol and generate data for publication, specifically Elyse Singer (NNAB), Debra Garcia (ACSR) and Salman Mahboob (ACSR). We would also like to acknowledge and thank Melissa Norström (formerly of Karolinska Institutet) for initially teaching us the HIV-1 single genome sequencing technique for the gag gene. This method adapted from protocols from previous publications (Palmer et al., 2005; Norström et al., 2012; Lamers et al., 2015; Rife et al., 2016).
References
- Abbas, W., Tariq, M., Iqbal, M., Kumar, A. and Herbein, G. (2015). Eradication of HIV-1 from the macrophage reservoir: an uncertain goal? Viruses 7(4): 1578-1598.
- Anderson, J. A., Archin, N. M., Ince, W., Parker, D., Wiegand, A., Coffin, J. M., Kuruc, J., Eron, J., Swanstrom, R. and Margolis, D. M. (2011). Clonal sequences recovered from plasma from patients with residual HIV-1 viremia and on intensified antiretroviral therapy are identical to replicating viral RNAs recovered from circulating resting CD4+ T cells. J Virol 85(10): 5220-5223.
- Autran, B., Carcelain, G., Li, T. S., Blanc, C., Mathez, D., Tubiana, R., Katlama, C., Debre, P. and Leibowitch, J. (1997). Positive effects of combined antiretroviral therapy on CD4+ T cell homeostasis and function in advanced HIV disease. Science 277(5322): 112-116.
- Bednar, M. M., Hauser, B. M., Zhou, S., Jacobson, J. M., Eron, J. J., Jr., Frank, I. and Swanstrom, R. (2016). Diversity and tropism of HIV-1 rebound virus populations in plasma level after treatment discontinuation. J Infect Dis 214(3): 403-407.
- Boritz, E. A., Darko, S., Swaszek, L., Wolf, G., Wells, D., Wu, X., Henry, A. R., Laboune, F., Hu, J., Ambrozak, D., Hughes, M. S., Hoh, R., Casazza, J. P., Vostal, A., Bunis, D., Nganou-Makamdop, K., Lee, J. S., Migueles, S. A., Koup, R. A., Connors, M., Moir, S., Schacker, T., Maldarelli, F., Hughes, S. H., Deeks, S. G. and Douek, D. C. (2016). Multiple origins of virus persistence during natural control of HIV infection. Cell 166(4): 1004-1015.
- Brown, R. J., Peters, P. J., Caron, C., Gonzalez-Perez, M. P., Stones, L., Ankghuambom, C., Pondei, K., McClure, C. P., Alemnji, G., Taylor, S., Sharp, P. M., Clapham, P. R. and Ball, J. K. (2011). Intercompartmental recombination of HIV-1 contributes to env intrahost diversity and modulates viral tropism and sensitivity to entry inhibitors. J Virol 85(12): 6024-6037.
- Buzon, M. J., Sun, H., Li, C., Shaw, A., Seiss, K., Ouyang, Z., Martin-Gayo, E., Leng, J., Henrich, T. J., Li, J. Z., Pereyra, F., Zurakowski, R., Walker, B. D., Rosenberg, E. S., Yu, X. G. and Lichterfeld, M. (2014). HIV-1 persistence in CD4+ T cells with stem cell-like properties. Nat Med 20(2): 139-142.
- Chun, T. W., Carruth, L., Finzi, D., Shen, X., DiGiuseppe, J. A., Taylor, H., Hermankova, M., Chadwick, K., Margolick, J., Quinn, T. C., Kuo, Y. H., Brookmeyer, R., Zeiger, M. A., Barditch-Crovo, P. and Siliciano, R. F. (1997). Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387(6629): 183-188.
- Chun, T. W., Finzi, D., Margolick, J., Chadwick, K., Schwartz, D. and Siliciano, R. F. (1995). In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat Med 1(12): 1284-1290.
- Crooks, A. M., Bateson, R., Cope, A. B., Dahl, N. P., Griggs, M. K., Kuruc, J. D., Gay, C. L., Eron, J. J., Margolis, D. M., Bosch, R. J. and Archin, N. M. (2015). Precise quantitation of the latent HIV-1 reservoir: Implications for eradication strategies. J Infect Dis 212(9): 1361-1365.
- Eriksson, S., Graf, E. H., Dahl, V., Strain, M. C., Yukl, S. A., Lysenko, E. S., Bosch, R. J., Lai, J., Chioma, S., Emad, F., Abdel-Mohsen, M., Hoh, R., Hecht, F., Hunt, P., Somsouk, M., Wong, J., Johnston, R., Siliciano, R. F., Richman, D. D., O'Doherty, U., Palmer, S., Deeks, S. G. and Siliciano, J. D. (2013). Comparative analysis of measures of viral reservoirs in HIV-1 eradication studies. PLoS Pathog 9(2): e1003174.
- Etemad, B., Ghulam-Smith, M., Gonzalez, O., White, L. F. and Sagar, M. (2015). Single genome amplification and standard bulk PCR yield HIV-1 envelope products with similar genotypic and phenotypic characteristics. J Virol Methods 214: 46-53.
- Evering, T. H., Kamau, E., St Bernard, L., Farmer, C. B., Kong, X. P. and Markowitz, M. (2014). Single genome analysis reveals genetic characteristics of Neuroadaptation across HIV-1 envelope. Retrovirology 11: 65.
- Finzi, D., Hermankova, M., Pierson, T., Carruth, L. M., Buck, C., Chaisson, R. E., Quinn, T. C., Chadwick, K., Margolick, J., Brookmeyer, R., Gallant, J., Markowitz, M., Ho, D. D., Richman, D. D. and Siliciano, R. F. (1997). Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278(5341): 1295-1300.
- Ho, Y. C., Shan, L., Hosmane, N. N., Wang, J., Laskey, S. B., Rosenbloom, D. I., Lai, J., Blankson, J. N., Siliciano, J. D. and Siliciano, R. F. (2013). Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 155(3): 540-551.
- Jordan, M. R., Kearney, M., Palmer, S., Shao, W., Maldarelli, F., Coakley, E. P., Chappey, C., Wanke, C. and Coffin, J. M. (2010). Comparison of standard PCR/cloning to single genome sequencing for analysis of HIV-1 populations. J Virol Methods 168(1-2): 114-120.
- Josefsson, L., von Stockenstrom, S., Faria, N. R., Sinclair, E., Bacchetti, P., Killian, M., Epling, L., Tan, A., Ho, T., Lemey, P., Shao, W., Hunt, P. W., Somsouk, M., Wylie, W., Douek, D. C., Loeb, L., Custer, J., Hoh, R., Poole, L., Deeks, S. G., Hecht, F. and Palmer, S. (2013). The HIV-1 reservoir in eight patients on long-term suppressive antiretroviral therapy is stable with few genetic changes over time. Proc Natl Acad Sci U S A 110(51): E4987-4996.
- Kandathil, A. J., Sugawara, S. and Balagopal, A. (2016). Are T cells the only HIV-1 reservoir? Retrovirology 13(1): 86.
- Kearney, M. F., Spindler, J., Shao, W., Yu, S., Anderson, E. M., O'Shea, A., Rehm, C., Poethke, C., Kovacs, N., Mellors, J. W., Coffin, J. M. and Maldarelli, F. (2014). Lack of detectable HIV-1 molecular evolution during suppressive antiretroviral therapy. PLoS Pathog 10(3): e1004010.
- Kearney, M. F., Wiegand, A., Shao, W., Coffin, J. M., Mellors, J. W., Lederman, M., Gandhi, R. T., Keele, B. F. and Li, J. Z. (2015). Origin of rebound plasma HIV includes cells with identical proviruses that are transcriptionally active before stopping of antiretroviral therapy. J Virol 90(3): 1369-1376.
- Lamers, S. L., Nolan, D. J., Rife, B. D., Fogel, G. B., McGrath, M. S., Burdo, T. H., Autissier, P., Williams, K. C., Goodenow, M. M. and Salemi, M. (2015). Tracking the emergence of host-specific simian immunodeficiency virus env and nef populations reveals nef early adaptation and convergent evolution in brain of naturally progressing rhesus macaques. J Virol 89(16): 8484-8496.
- Lamers, S. L, Rose, R., Maidji, E., Agsalda-Garcia, M., Nolan, D. J., Fogel, G. B., Salemi, M., Garcia, D. L., Bracci, P., Yong, W., Commins, D., Said, J., Khanlou, N., Hinkin, C. H., Sueiras, M. V., Mathisen, G., Donovan, S., Shiramizu, B., Stoddart, C. A., McGrath, M. S. and Singer, E. J. (2016a). HIV DNA is frequently present within pathologic tissues evaluated at autopsy from combined antiretroviral therapy-treated patients with undetectable viral loads. J Virol 90(20): 8968-83.
- Lamers, S. L., Rose, R., Nolan, D. J., Fogel, G. B., Barbier, A. E., Salemi, M. and McGrath, M. S. (2016b). HIV-1 evolutionary patterns associated with metastatic Kaposi's sarcoma during AIDS. Sarcoma 2016: 4510483.
- Iyer, S., Casey, E., Bouzek, H., Kim, M., Deng, W., Larsen, B. B., Zhao, H., Bumgarner, R. E., Rolland, M. and Mullins, J. I. (2015). Comparison of major and minor viral SNPs identified through single template sequencing and pyrosequencing in acute HIV-1 infection. PLoS One 10(8): e0135903.
- Norström M. M., Buggert, M., Tauriainen, J., Hartogensis, W., Prosperi, M. C., Wallet, M. A., Hecht, F. M., Salemi, M. and Karlsson, A. C. (2012). Combination of immune and viral factors distinguishes low-risk versus high-risk HIV-1 disease progression in HLA-B*5701 subjects. J Virol 86(18): 9802-9816.
- Novitsky, V., Wang, R., Rossenkhan, R., Moyo, S. and Essex, M. (2013). Intra-host evolutionary rates in HIV-1C env and gag during primary infection. Infect Genet Evol 19: 361-368.
- Palmer, S., Kearney, M., Maldarelli, F., Halvas, E. K., Bixby, C. J., Bazmi, H., Rock, D., Falloon, J., Davey, R. T., Jr., Dewar, R. L., Metcalf, J. A., Hammer, S., Mellors, J. W. and Coffin, J. M. (2005). Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J Clin Microbiol 43(1): 406-413.
- Palmer, S., Shafer, R. W. and Merigan, T. C. (1999). Highly drug-resistant HIV-1 clinical isolates are cross-resistant to many antiretroviral compounds in current clinical development. AIDS 13(6): 661-667.
- Rife, B. D., Nolan, D. J., Lamers, S. L., Autissier, P., Burdo, T., Williams, K. C. and Salemi, M. (2016). Evolution of neuroadaptation in the periphery and purifying selection in the brain contribute to compartmentalization of simian immunodeficiency virus (SIV) in the brains of rhesus macaques with SIV-associated encephalitis. J Virol 90(13): 6112-6126.
- Rodrigo, A. G., Goracke, P. C., Rowhanian, K. and Mullins, J. I. (1997). Quantitation of target molecules from polymerase chain reaction-based limiting dilution assays. AIDS Res Hum Retroviruses 13(9): 737-742.
- Rose, R., Lamers, S. L., Nolan, D. J., Maidji, E., Faria, N. R., Pybus, O. G., Dollar, J. J., Maruniak, S. A., McAvoy, A. C., Salemi, M., Stoddart, C. A., Singer, E. J. and McGrath, M. S. (2016). HIV maintains an evolving and dispersed population in multiple tissues during suppressive combined antiretroviral therapy in individuals with cancer. J Virol 90(20): 8984-8993.
- Rothenberger, M. K., Keele, B. F., Wietgrefe, S. W., Fletcher, C. V., Beilman, G. J., Chipman, J. G., Khoruts, A., Estes, J. D., Anderson, J., Callisto, S. P., Schmidt, T. E., Thorkelson, A., Reilly, C., Perkey, K., Reimann, T. G., Utay, N. S., Nganou Makamdop, K., Stevenson, M., Douek, D. C., Haase, A. T. and Schacker, T. W. (2015). Large number of rebounding/founder HIV variants emerge from multifocal infection in lymphatic tissues after treatment interruption. Proc Natl Acad Sci U S A 112(10): E1126-1134.
- Sacha, J. B. and Ndhlovu, L. C. (2016). Strategies to target non-T-cell HIV reservoirs. Curr Opin HIV AIDS 11(4): 376-382.
- Salemi, M. and Rife, B. (2016). Phylogenetics and phyloanatomy of HIV/SIV intra-host compartments and reservoirs: the key role of the central nervous system. Curr HIV Res 14(2):110-20.
- Sanborn, K. B., Somasundaran, M., Luzuriaga, K. and Leitner, T. (2015). Recombination elevates the effective evolutionary rate and facilitates the establishment of HIV-1 infection in infants after mother-to-child transmission. Retrovirology 12: 96.
- Shafer, R. W., Warford, A., Winters, M. A. and Gonzales, M. J. (2000). Reproducibility of human immunodeficiency virus type 1 (HIV-1) protease and reverse transcriptase sequencing of plasma samples from heavily treated HIV-1-infected individuals. J Virol Methods 86(2): 143-153.
- Simonetti, F. R., Sobolewski, M. D., Fyne, E., Shao, W., Spindler, J., Hattori, J., Anderson, E. M., Watters, S. A., Hill, S., Wu, X., Wells, D., Su, L., Luke, B. T., Halvas, E. K., Besson, G., Penrose, K. J., Yang, Z., Kwan, R. W., Van Waes, C., Uldrick, T., Citrin, D. E., Kovacs, J., Polis, M. A., Rehm, C. A., Gorelick, R., Piatak, M., Keele, B. F., Kearney, M. F., Coffin, J. M., Hughes, S. H., Mellors, J. W. and Maldarelli, F. (2016). Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc Natl Acad Sci U S A 113(7): 1883-1888.
- Sturdevant, C. B., Dow, A., Jabara, C. B., Joseph, S. B., Schnell, G., Takamune, N., Mallewa, M., Heyderman, R. S., Van Rie, A. and Swanstrom, R. (2012). Central nervous system compartmentalization of HIV-1 subtype C variants early and late in infection in young children. PLoS Pathog 8(12): e1003094.
- Svicher, V., Ceccherini-Silberstein, F., Antinori, A., Aquaro, S. and Perno, C. F. (2014). Understanding HIV compartments and reservoirs. Curr HIV/AIDS Rep 11(2): 186-194.
- Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M. and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10): 2731-2739.
- Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F. and Higgins, D. G. (1997). The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25(24): 4876-4882.
- Wagner, T. A., McKernan, J. L., Tobin, N. H., Tapia, K. A., Mullins, J. I. and Frenkel, L. M. (2013). An increasing proportion of monotypic HIV-1 DNA sequences during antiretroviral treatment suggests proliferation of HIV-infected cells. J Virol 87(3): 1770-1778.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Nolan, D. J., Lamers, S. L., Rose, R., Dollar, J. J., Salemi, M. and McGrath, M. S. (2017). Single Genome Sequencing of Expressed and Proviral HIV-1 Envelope Glycoprotein 120 (gp120) and nef Genes. Bio-protocol 7(12): e2334. DOI: 10.21769/BioProtoc.2334.
Category
Microbiology > Microbial genetics > DNA > DNA sequencing
Microbiology > Microbial genetics > RNA > RNA extraction
Molecular Biology > DNA > Genotyping
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.