- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Fluorescently Labelled Aerolysin (FLAER) Labelling of Candida albicans Cells
Published: Vol 7, Iss 11, Jun 5, 2017 DOI: 10.21769/BioProtoc.2303 Views: 10849
Reviewed by: Yanjie LiEmmanuel ZavalzaMichael Tscherner
Original research article
The authors used this protocol in:

Aug 2016
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
In this protocol we describe a nonradiolabelled labelling of GPI anchor in Candida albicans. The method uses a fluorescent probe to bind specifically to GPI anchors so that the level of GPI-anchored proteins at the cell surface can be measured. The labelling does not need permeabilization of cells and can be carried out in vivo.
Keywords: FLAERBackground
The GPI (glycosylphosphatidylinositol) anchor is a post translational modification occurring in the endoplasmic reticulum (ER). The preformed GPI anchor is attached in the lumen of the ER to the C-terminus of specific proteins that carry a GPI anchor attachment signal sequence. These proteins are subsequently transported (and extensively further modified) by the secretory pathway to the cell surface where the proteins are present anchored to the extracellular leaflet of the plasma membrane or covalently linked to the cell wall in organisms that have a wall. A variety of proteins get GPI anchored proteins in eukaryotes, for example, hydrolytic enzymes, cell surface adhesion molecules or receptor proteins (Orlean and Menon, 2007). In fungal pathogens, such as Candida albicans, many of the adhesins involved in host recognition and adherence, as well as several pathogenesis and virulence factors are GPI anchored proteins; besides, several GPI-anchored proteins in Candida albicans have proteolytic activity (Richard and Plaine, 2007; Nobile et al., 2008). The pathway is essential in Candida albicans and downregulating it or targeting it could be a useful strategy to combat Candida infections. The protocol used for assessing GPI anchor levels in Candida albicans is elaborated here.
Fluorescently Labelled Aerolysin (FLAER) is a fluorescein labelled inactive derivative of aerolysin, a Gram-negative bacterial toxin, which binds to GPI-anchored proteins in the cell membrane of the eukaryotic host and forms pores (Howard et al., 1987; Parker et al., 1994). In FLAER, proaerolysin is tagged with Alexa Fluor 488 dye which can bind to GPI anchors of eukaryotic cells without any harm to the cell (Sutherland et al., 2007). FLAER is also clinically used to detect GPI anchor levels in Paroxysmal Nocturnal Hemoglobinuria (PNH) cells, a disease caused by somatic mutations in PIGA (a subunit of glycosylphosphatidylinositol-N-acetylglucosamine transferase enzyme complex) in mammals (Brodsky et al., 2000). Once stained with FLAER, the fluorescence of cells can be monitored/quantified under a confocal fluorescence microscope in the GFP channel and the data used as a measure of GPI anchored proteins on the cell surface. In a previous study, we monitored the levels of GPI-anchored proteins on the cell surface of a GPI biosynthetic mutant, Cagpi14, in Candida albicans by using FLAER (Singh et al., 2016). CaGPI14 encodes for the catalytic subunit of the first mannosyltransferase of the GPI-anchor biosynthesis pathway. Deficiency in CaGpi14 severely affects cell growth and wall integrity in the organism although low levels of expression appear to be sufficient to make the cells viable. Its homolog in S. cerevisiae is essential (Kim et al., 2007). The protocol for FLAER labelling used to assess GPI anchor levels in this mutant of Candida albicans is described here.
Materials and Reagents
- Pipette tips
- Sterile plastic toothpick
- Culture tubes (50 ml)
- 1.5 ml micro-centrifuge tubes
- Glass slides
- Cover slips
- Candida albicans strains CAF2-1 (URA3/ura3::imm434 IRO1/iro1::imm434) and conditional null mutant of CaGPI14 (CAF2-1::Cagpi14/pMET3-CaGPI14), encoding the catalytic subunit of the first mannosyltransferase in the GPI biosynthetic pathway, for this study
- Synthetic defined (SD) medium with Uridine+Cys-Met- dropout mix was made in the lab for this study since the strains were uridine auxotrophs. Cys/Met was used to repress CaGPI14 expression (all L-amino acids were obtained from Sisco Research Laboratory). This was possible because the conditional mutant strain had the only surviving allele of CaGPI14 placed under the control of the MET3 promoter, which permits gene expression in the absence of Cys/Met and represses its expression in the presence of Cys/Met
- Liquid FLAER (25 µg/0.5 ml) from Pinewood Scientific Services Inc. (FL1S-C)
- 50% glycerol (Merck, catalog number: 1.07051.0521 )
- D-glucose (Fisher Scientific, catalog number: D16-500 )
- Yeast nitrogen base (HiMedia Laboratories, catalog number: M878 )
- Cysteine (Sisco Research Laboratory, catalog number: 034890 )
- Methionine (Sisco Research Laboratory, catalog number: 134869 )
- Sodium chloride (NaCl) (Fisher Scientific, catalog number: 15915 )
- Potassium chloride (KCl) (Merck, catalog number: 1049360500 )
- Sodium phosphate dibasic (Na2HPO4) (Fisher Scientific, catalog number: S374-3 )
- Potassium phosphate monobasic (KH2PO4) (Merck, catalog number: 1.93205.0521 )
- Hydrochloric acid (HCl)
- SD Uri+Cys-Met-/SD Uri+Cys+Met+ broth (100 ml) (see Recipes)
- Phosphate buffered saline (PBS; pH 7.5) (see Recipes)
Equipment
- Pipettes
100-1,000 µl (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 4642090 )
20-200 µl (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 4642080 )
2-20 µl (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 4642060 ) - Vortex mixer (REMI GROUP, model: CM-101 )
- Shaker incubator (Set at either 30 °C or 20 °C according to requirement) (Labtech, model: LSI-5002M )
- Centrifuge (Plasto Craft Industries, model: Micro-spin R-V/FM )
- Spectrophotometer (Thermo Fisher Scientific, Thermo ScientificTM, model: MultiskanTM GO Microplate Spectrophotometer , catalog number: 51119300)
- Andor spinning Disk confocal microscope
- Autoclave
Software
- Andor iQ2.7 (for image capturing)
- GIMP 2.8.18 software (for quantification)
- Origin 0.5 software
Procedure
- Primary culture of all the strains is set up in 10 ml of SD Uri+Cys-Met- medium using a tip of a stock culture picked up with a sterile plastic toothpick. The cultures are allowed to grow at 30 °C overnight in shaking condition at 220 rpm.
- Secondary cultures are set up with 2% of overnight grown cultures (200 μl of overnight grown culture in 10 ml) in fresh appropriate medium (CAF2-1 in SD Uri+Cys-Met- and conditional null of CaGPI14 in SD Uri+Cys-Met- [permissive] as well as in SD Uri+Cys+Met+ [repressive] media) at 30 °C with shaking (220 rpm).
- After 5 h of growth (log phase) the cells are spun down by centrifugation at 3,000 x g for 5 min at room temperature (30 °C). Cells in log phase are preferred since we observed extensive cell clumping in the Cagpi14 mutant cells at late stages of growth and in stationary phase. It is also important to point out that in saturated cultures, cell death processes may be initiated and this in turn alters cell wall and membrane stability, affecting the staining of the cells. To avoid errors arising from these problems, log phase cells are preferred.
- The cell pellets are washed with PBS by centrifugation at 3,000 x g and resuspended in 10 ml of PBS.
- Equal numbers of cells in each case, corresponding to OD600 nm ~0.6, are taken and resuspended in a total volume of 200 µl PBS in 1.5 ml micro-centrifuge tubes.
- FLAER (10 μl of 50 μg/ml i.e., 0.5 μg) is added to the 200 μl cell suspension (1:20 dilution).
- After mild vortexing at ~500 rpm, the cell suspension is incubated for 1 h in a shaker-incubator (70 rpm) at 20 °C in the dark.
- The cells are then spun down at 3,000 x g for 3 min and the cell pellets washed thrice with PBS by pipetting.
- For microscopic analysis cell pellets are resuspended in 40 µl of 50% glycerol (v/v in water).
- 5 µl of cell suspensions are spotted on glass slides and covered with cover slips.
- Fluorescent images of the cells are captured using a confocal fluorescence microscope with an Alexa 488 filter with 0.4 sec and 0.2 sec exposure time for fluorescence and DIC images, respectively. Camera EM gain is 100 with camera exposure units of 1 for both type of images. A representative image for the obtained results is shown in Figure 1.
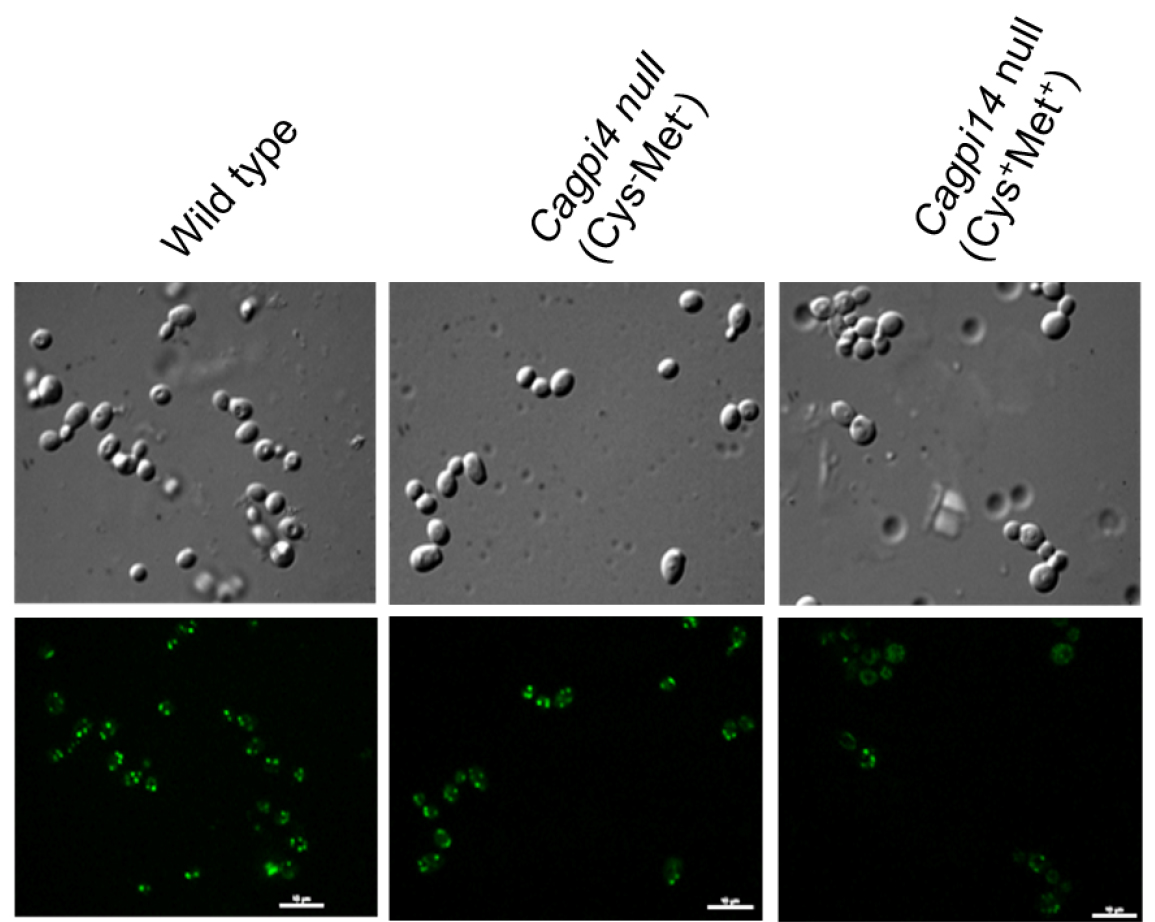
Figure 1. Confocal imaging of FLAER labelled cells. Images of wild type (CAF2-1) and Cagpi14 conditional null mutant cells under permissive (Cys-Met-) as well as repressive (Cys+Met+) conditions after incubation with FLAER as described above. Scale bars = 10 µm in each image.
Data analysis
Quantification of fluorescence intensity:
- Fluorescence intensity of cells can be quantified using Nikon-AR-analysis or GIMP software. For each strain fluorescence intensity of at least 50 cells are counted. For the data plotted here, quantification was done using GIMP software. Schematic representation of quantification is shown in Figure 2. P-values for statistical significance of data are calculated by using the Student’s t-test in Microsoft Excel.
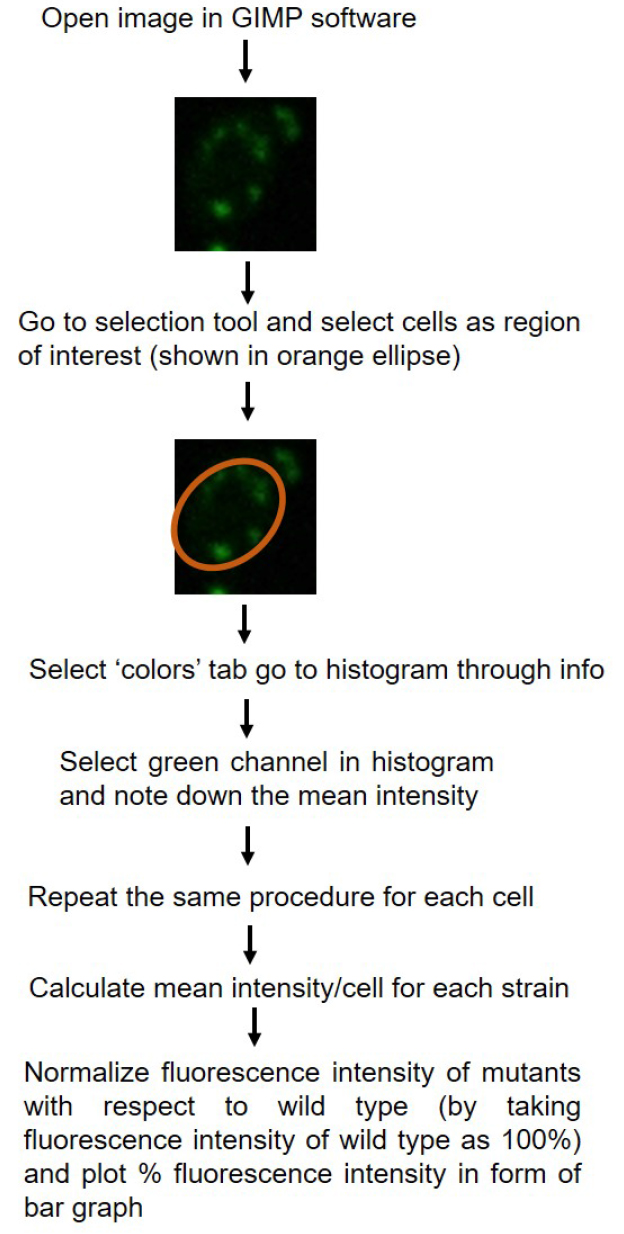
Figure 2. Schematic diagram showing quantification of fluorescence intensity using GIMP 2.8.18 software. Quantification of single fluorescent cells has been shown as example. - Relative intensity for the mutant is plotted with reference to the fluorescence intensity of wild type strain (CAF2-1) taken as 100%.
- The data points are plotted in Origin 0.5 software (Figure 3) in form of bar graph.
- In the above experiment we find roughly 40% lower fluorescence in the conditional null mutant (under repressive conditions) as compared to the wild type strain, CAF2-1. Under permissive conditions, only 20% reduction in fluorescence intensity is observed relative to the wild type strain.
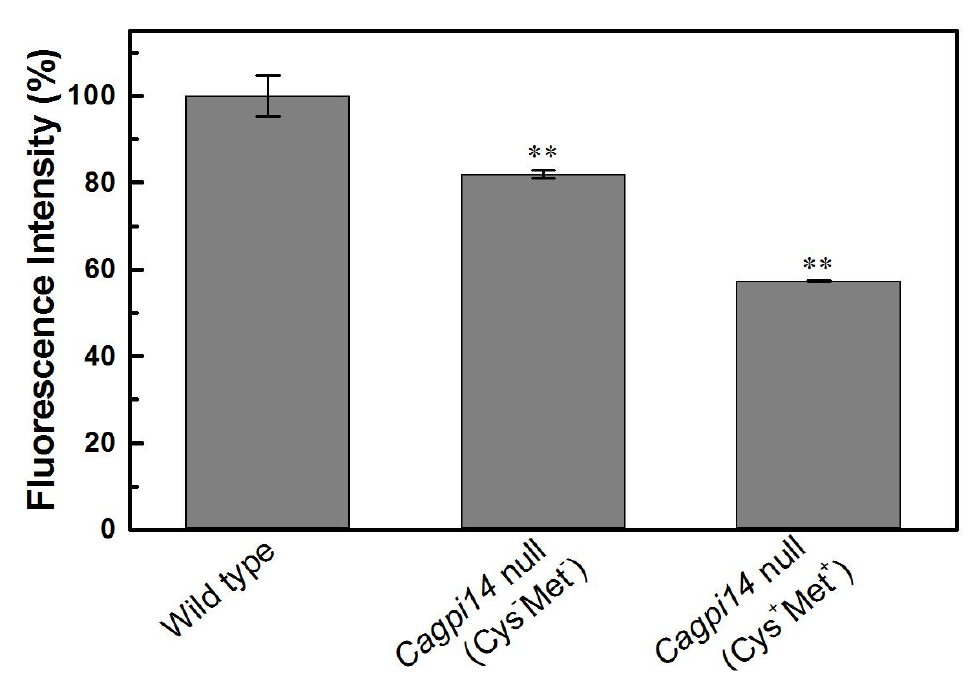
Figure 3. Quantification of the relative fluorescence intensity of FLAER labelled cells. The percent fluorescence intensity of the Cagpi14 conditional null mutant cells is plotted relative to that of wild type (CAF2-1) cells (100%). P-value < 0.003 (**).
Notes
- Labelling should be performed with cells from a secondary culture in the log-phase of growth.
- Conditions that promote clumping of cells should be avoided as this introduces errors in estimation of cell numbers as well as in levels of staining.
- Images should be captured on the same day of the labelling.
Recipes
- SD Uri+Cys-Met-/SD Uri+Cys+Met+ broth (100 ml)
2 g D-glucose
0.67 g YNB
0.2 g Uri-Cys-Met- dropout mix (Mixture of all L-amino acids except L-cysteine, L-methionine and uridine; 1 g of each amino acid except for adenine [0.25 g] and para-aminobenzoic acid [0.1 g])
1 M uridine solution (made in autoclaved water)
Add above chemicals in 80 ml double distilled water and dissolve
Add 500 μl of 1 M uridine solution to it and make up the volume to 100 ml
For making SD Uri+Cys+Met+ medium add cysteine (5 mM) and methionine (5 mM) to SD Uri+Cys-Met- medium and autoclave - Phosphate buffer saline (PBS), pH 7.5 (1 L)
8 g NaCl
0.2 g KCl
1.44 g Na2HPO4
0.24 g KH2PO4
Dissolve the above chemicals in 800 ml double distilled water
Adjust pH to 7.5 with HCl
Make up the volume with double distilled water
Acknowledgments
The above protocol is adapted from our previous study (Singh et al., 2016). The work has been funded by a senior research fellowship from Indian Council of Medical Research (ICMR) to SLS and a research grant (No. SB/OC/CB-03A/2014) from Department of Science and Technology (DST), India to SSK.
References
- Brodsky, R. A., Mukhina, G. L., Li, S., Nelson, K. L., Chiurazzi, P. L., Buckley, J. T. and Borowitz, M. J. (2000). Improved detection and characterization of paroxysmal nocturnal hemoglobinuria using fluorescent aerolysin. Am J Clin Pathol 114(3): 459-466.
- Howard, S. P., Garland, W. J., Green, M. J. and Buckley, J. T. (1987). Nucleotide sequence of the gene for the hole-forming toxin aerolysin of Aeromonas hydrophila. J Bacteriol 169(6): 2869-2871.
- Kim, Y.U., Ashida, H., Mori, K., Maeda, Y., Hong, Y. and Kinoshita, T. (2007). Both mammalian PIG-M and PIG-X are required for growth of GPI14-disrupted yeast. J Biochem 142(1): 123-129.
- Nobile, C. J., Schneider, H. A., Nett, J. E., Sheppard, D. C., Filler, S. G., Andes, D. R. and Mitchell, A. P. (2008). Complementary adhesin function in C. albicans biofilm formation. Curr Biol 18(14): 1017-1024.
- Orlean, P. and Menon, A. K. (2007). Thematic review series: lipid posttranslational modifications. GPI anchoring of protein in yeast and mammalian cells, or: how we learned to stop worrying and love glycophospholipids. J Lipid Res 48(5): 993-1011.
- Parker, M. W., Buckley, J. T., Postma, J. P., Tucker, A. D., Leonard, K., Pattus, F. and Tsernoglou, D. (1994). Structure of the Aeromonas toxin proaerolysin in its water-soluble and membrane-channel states. Nature 367(6460): 292-295.
- Richard, M. L. and Plaine, A. (2007). Comprehensive analysis of glycosylphosphatidylinositol-anchored proteins in Candida albicans. Eukaryot Cell 6(2): 119-133.
- Singh, S. L., Rai, R. C., Sah, S. K. and Komath, S. S. (2016). The catalytic subunit of the first mannosyltransferase in the GPI biosynthetic pathway affects growth, cell wall integrity and hyphal morphogenesis in Candida albicans. Yeast 33(8): 365-383.
- Sutherland, D. R., Kuek, N., Davidson, J., Barth, D., Chang, H., Yeo, E., Bamford, S., Chin-Yee, I. and Keeney, M. (2007). Diagnosing PNH with FLAER and multiparameter flow cytometry. Cytometry B Clin Cytom 72(3): 167-177.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Singh, S. L. and Komath, S. S. (2017). Fluorescently Labelled Aerolysin (FLAER) Labelling of Candida albicans Cells. Bio-protocol 7(11): e2303. DOI: 10.21769/BioProtoc.2303.
Category
Microbiology > Microbial cell biology > Cell imaging
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.