- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Evaluation of Plasmid Stability by Negative Selection in Gram-negative Bacteria


Published: Vol 7, Iss 9, May 5, 2017 DOI: 10.21769/BioProtoc.2261 Views: 12912
Reviewed by: Valentine V TrotterAlba BlesaSeda Ekici
Original research article
The authors used this protocol in:

14-Oct 2013
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Plasmid stability can be measured using antibiotic-resistance plasmid derivatives by positive selection. However, highly stable plasmids are below the sensitivity range of these assays. To solve this problem we describe a novel, highly sensitive method to measure plasmid stability based on the selection of plasmid-free cells following elimination of plasmid-containing cells. The assay proposed here is based on an aph-parE cassette. When synthesized in the cell, the ParE toxin induces cell death. ParE synthesis is controlled by a rhamnose-inducible promoter. When bacteria carrying the aph-parE module are grown in media containing rhamnose as the only carbon source, ParE is synthesized and plasmid-containing cells are eliminated. Kanamycin resistance (aph) is further used to confirm the absence of the plasmid in rhamnose grown bacteria.
Keywords: Plasmid stabilityBackground
lassically, plasmid stability has been measured by positive selection using antibiotic-resistance plasmid derivatives. Cells harbouring the studied plasmid are positively selected in the presence of the selection antibiotic (Gerdes et al., 1985; del Solar et al., 1987). The main drawback of this technique is its sensitivity; highly stable plasmids are below the sensitivity of these assays. To solve this problem alternative methods relying on the direct selection of plasmid-free cells such as the tetAR-chlortetracycline system, have been described (Bochner et al., 1980; Maloy and Nunn, 1981; Garcia-Quintanilla et al., 2006). Limitations of the tetAR-chlortetracycline method include poor reproducibility and the frequent occurrence of false positives (Li et al., 2013). Here, we describe a novel, highly sensitive plasmid stability assay based on the counter-selection of plasmid-containing cells. This assay is based on a cassette containing a ParE toxin-encoding gene controlled by a rhamnose-inducible promoter and a kanamycin resistance gene (aph) (Figure 1) (Maisonneuve et al., 2011). ParE is the toxin of the toxin-antitoxin system parDE, and targets the DNA gyrase, blocks DNA replication and induces DNA breaks leading to cell death (Jiang et al., 2002). The aph-parE cassette is inserted into the plasmid of interest using homologous recombination. Upon induction of PparE in minimal media containing rhamnose as the only carbon source, only plasmid-free cells survive (Lobato-Marquez et al., 2016). Kanamycin is then used to confirm the loss of the plasmid (Figure 2).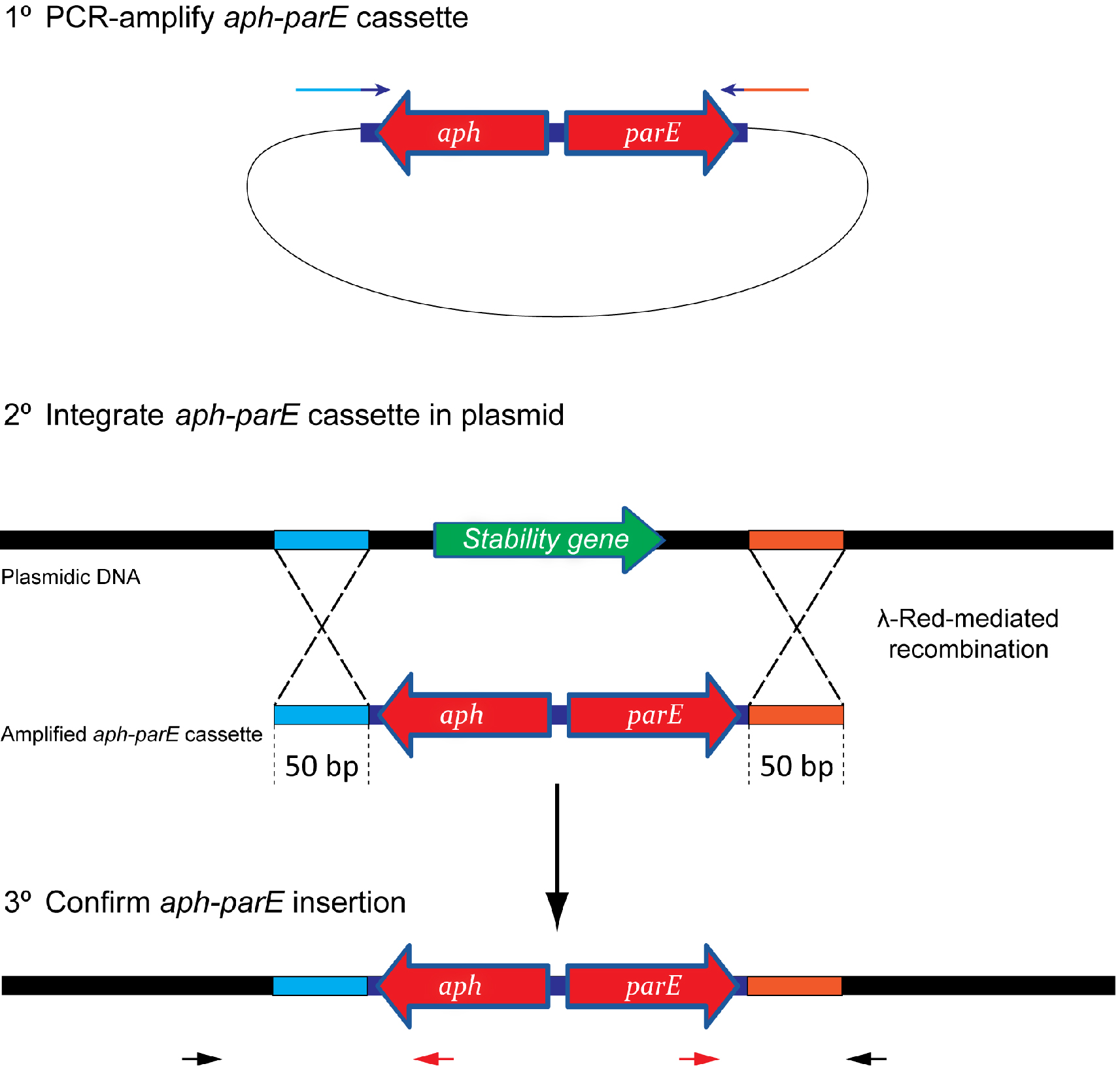
Figure 1. Scheme showing the integration process of the aph-parE cassette into the plasmid of interest. (1) The aph-parE cassette is first amplified by PCR using pKD267 plasmid as template. (2) Then, cells harbouring a plasmid encoding λ-Red recombinase are electroporated with aph-parE DNA fragment. λ-Red recombinase directs the specific integration of the aph-parE module into the plasmid region containing the 50 bp upstream and 50 bp downstream homologous sequences included in the oligos used for PCR. (3) Confirm the aph-parE insertion by using primers annealing with the cassette (red arrows) and with the plasmid (black arrows).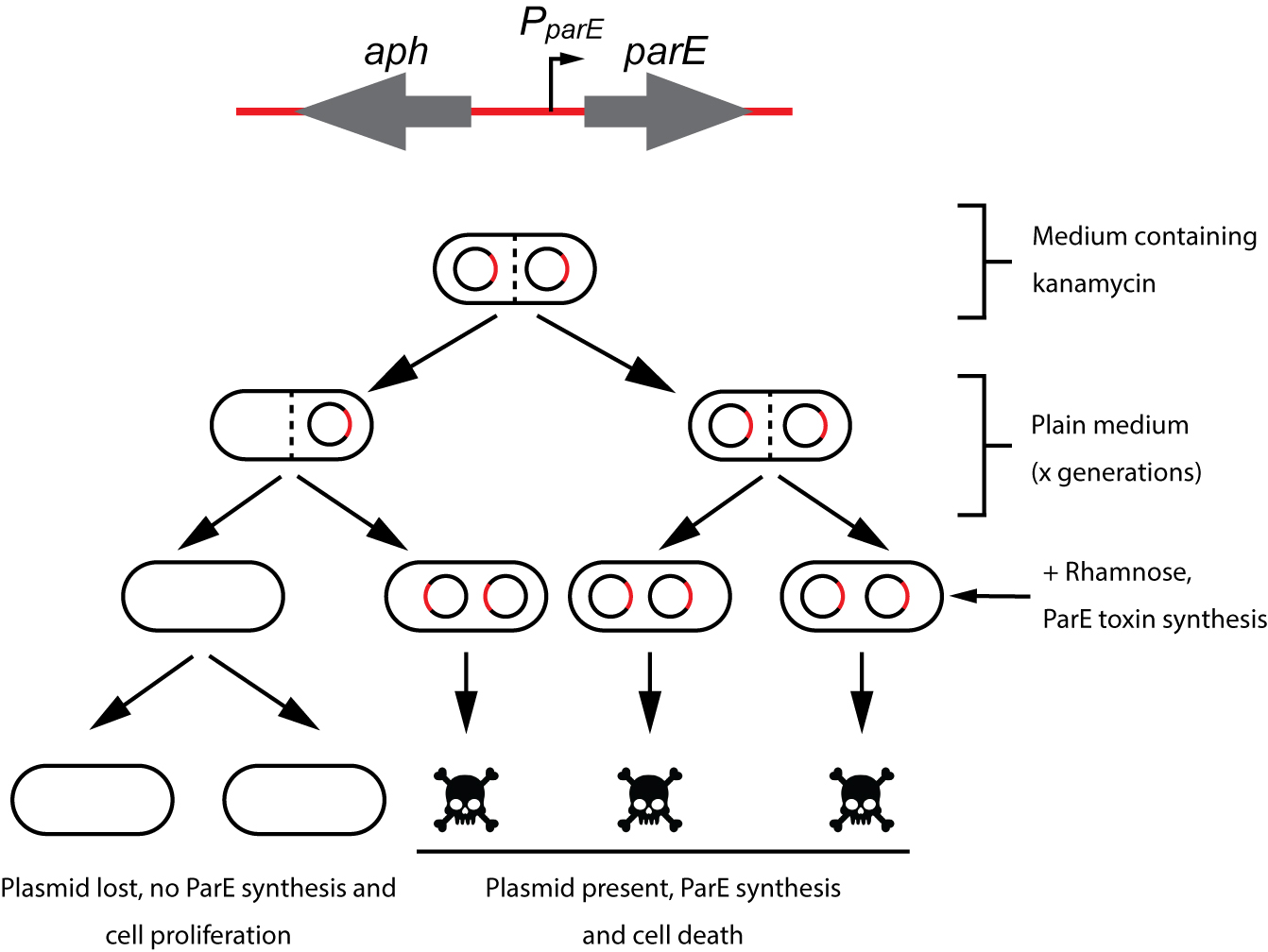
Figure 2. Plasmid stability procedure. To avoid plasmid loss, the strain carrying the aph-parE cassette is initially grown under antibiotic selection pressure (using kanamycin). When kanamycin is removed from the medium, the plasmid of interest will be lost after a certain number of generations. Plasmid-free cells are selected when the culture is plated in M9-rhamnose plates containing rhamnose as the only carbon source. Modified from Lobato-Marquez et al., 2016.
Materials and Reagents
- Pipette tips
- 1.5 ml Eppendorf tubes
- Millipore 0.22 µm pore size filter (EMD Millipore, catalog number: SLGP033RS )
- 9-cm sterile Petri dishes
- Electroporation cuvettes 0.2 cm gap (Bio-Rad Laboratories, catalog number: 1652082 )
- Glass beads for bacterial plating (VWR, catalog number: 201-0279 )
- pKD267 plasmid (described in Maisonneuve et al., 2011)
- pKD46 plasmid (described in Datsenko and Wanner, 2000)
- Expand High Fidelity DNA polymerase (Roche Molecular Systems, catalog number: 11732641001 )
- DpnI restriction enzyme (New England Biolabs, catalog number: R0176 )
- PCR purification kit (5Prime, catalog number: 2300610 )
- Plasmid purification kit (Roche Molecular Systems, catalog number: 11754777001 )
- 4 °C chilled sterile distilled MilliQ water
- Distilled MilliQ water
- Ampicillin sodium salt (Sigma-Aldrich, catalog number: A0166 )
- L-arabinose (Sigma-Aldrich, catalog number: A3256 )
- Kanamycin (Sigma-Aldrich, catalog number: K1876 )
- D-glucose (Sigma-Aldrich, catalog number: G8270 )
- Bacto tryptone (BD, BactoTM, catalog number: 211705 )
- Bacto yeast extract (BD, BactoTM, catalog number: 288620 )
- Sodium chloride (NaCl) (VWR, BDH®, catalog number: 102415K )
- European bacteriological agar (Conda, catalog number: 1800 )
- Sodium phosphate dibasic (Na2HPO4) (Sigma-Aldrich, catalog number: 71645 )
- Potassium phosphate monobasic (KH2PO4) (Sigma-Aldrich, catalog number: P9791 )
- Ammonium chloride (NH4Cl) (Sigma-Aldrich, catalog number: A9434 )
- Calcium chloride dihydrate (CaCl2·2H2O) (Sigma-Aldrich, catalog number: C3881 )
- Magnesium sulfate (MgSO4) (VWR, BDH®, catalog number: 291175X )
- Vitamin B1 (Thiamine) (Sigma-Aldrich, catalog number: T4625 )
- L-rhamnose monohydrate (Sigma-Aldrich, catalog number: R3875 )
- Potassium chloride (KCl)
- Na2HPO4·12H2O
- Glycerol (v/v) (Sigma-Aldrich, catalog number: G5516 )
- Luria-Bertani (LB) broth medium (see Recipes)
- Luria Bertani plates (see Recipes)
- M9 (10x) (see Recipes)
- CaCl2/MgSO4 solution (100x)
- M9 minimum medium (see Recipes)
- M9-rhamnose plates (see Recipes)
- Phosphate saline buffered (PBS) (see Recipes)
- 10% sterile glycerol (see Recipes)
- L-rhamnose (see Recipes)
Equipment
- Selection of single channel pipettes (2 µl, 20 µl, 20 µl, 1,000 µl) (Gilson, P-2, P-20, P-200, P-1000)
- Glass 100 ml flasks (Fisher Scientific, Fisherbrand)
- Glass 50 ml flasks (Fisher Scientific, Fisherbrand)
- MiniSpin® Eppendorf benchtop centrifuge (Eppendorf, model: MiniSpin® )
- Bacterial shaking incubator (Eppendorf, New Brunswick, model: Innova® 4000 )
- MicroPulserTM electroporator (Bio-Rad Laboratories, model: MicroPulser Electroporator , catalog number: 1652100)
- Eppendorf refrigerated centrifuge (Eppendorf, model: 5804 )
- Spectrophotometer (Thermo Fisher Scientific, Thermo ScientificTM, model: SPECTRONICTM 200 )
- Milli-Q® integral water purification system for ultrapure (Nanopure) water
- DNA SpeedVac (Savant Systems, model: SpeedVac DNA 110 )
- Autoclave (Prestige Medical, catalog number: 210004 )
Software
- GraphPad Prism Software (https://www.graphpad.com/scientific-software/prism/)
Procedure
This procedure is only designed to study the stability of plasmids carried by bacterial species able to uptake rhamnose from the medium, and species in which the ParE toxin is functional. This should be considered before trying to adapt this procedure to new species. Although we employed this assay to measure the stability of the virulence plasmid of Salmonella enterica subs. enterica serovar Typhimurium str. SV5015 (Lobato-Marquez et al., 2016), the method is also useful for E. coli plasmids.
- Design of recombinant plasmid variants
- Amplify aph-parE cassette by polymerase chain reaction (PCR) using pKD267 plasmid as template and Expand High Fidelity DNA polymerase (~1,800 bp) (see Notes 1 and 2, Table 1 and Figure 1). It is recommended to use the following PCR program: 94 °C 5 min, [94 °C 30 sec; 60 °C 30 sec; 72 °C 2 min] x 10, [94 °C 30 sec; 60 °C 30 sec; 72 °C 2 min + 5 sec increment per cycle] x 25 cycles.
- Digest the PCR-amplified aph-parE cassette by adding 1 µl of DpnI per 50 µl of PCR reaction. DpnI digests methylated DNA, thus eliminating the parental pKD267 plasmid but not the amplified aph-parE DNA module.
- Purify the DpnI-digested PCR reaction using a 5Prime PCR purification kit. Elute DNA in MilliQ water. Alternatively, standard precipitation procedures such as the phenol/chloroform precipitation method, can be used to purify the aph-parE DNA fragment (see Note 3).
- Concentrate purified-PCR product to 500-800 ng/µl using DNA SpeedVac.
- Transform the strain containing the plasmid for which the stability is to be analyzed, with pKD46 (see Note 4 and Figure 3). Grow the resulting variant in LB containing 50 µg/ml of ampicillin at 30 °C.
- Make electrocompetent cells of the strain carrying pKD46 (Figure 3). Pick one colony of the new strain carrying pKD46 plasmid and inoculate 5 ml of LB supplemented with 50 µg/ml of ampicillin and 0.4% arabinose (w/v). Grow the culture for 16 h at 30 °C in 10 ml LB in a 100 ml flask with shaking (~150 rpm).
- Dilute 1:100 of overnight culture in LB containing 50 µg/ml of ampicillin and 0.4% arabinose in 10:1 flask:medium volume ratio flask.
- Grow the bacterial culture at 30 °C and 150 rpm up to an optical density (measured at 600 nm) of 0.6.
- Centrifuge the bacterial culture in a 4 °C refrigerated centrifuge for 5 min at 15,557 x g.
- Discard the supernatant and rinse the bacterial pellet twice with 4 °C chilled sterile distilled water.
- Discard the supernatant and rinse the bacterial pellet with 4 °C sterile 10% glycerol.
- Resuspend the bacterial pellet in sterile 10% glycerol (500 µl per 50 ml of bacterial culture).
- Aliquot bacterial electrocompetent cells in 1.5 ml Eppendorf tubes (250 µl culture per tube). Keep on ice if competent cells are going to be used immediately. Otherwise, electrocompetent cells should be kept at -80 °C.
- Electroporate pKD46 competent cells (2.5 kV, 5 ms) with 800-1,000 ng of purified aph-parE DNA fragment (Figure 3) and grow cells for 3 h at 37 °C (this temperature promotes the loss of pKD46).
- Plate electroporated cells in LB-plates containing 50 µg/ml of kanamycin and 0.2% of glucose (see Note 5).
- Confirm the proper integration of the aph-parE cassette in your plasmid using PCR.
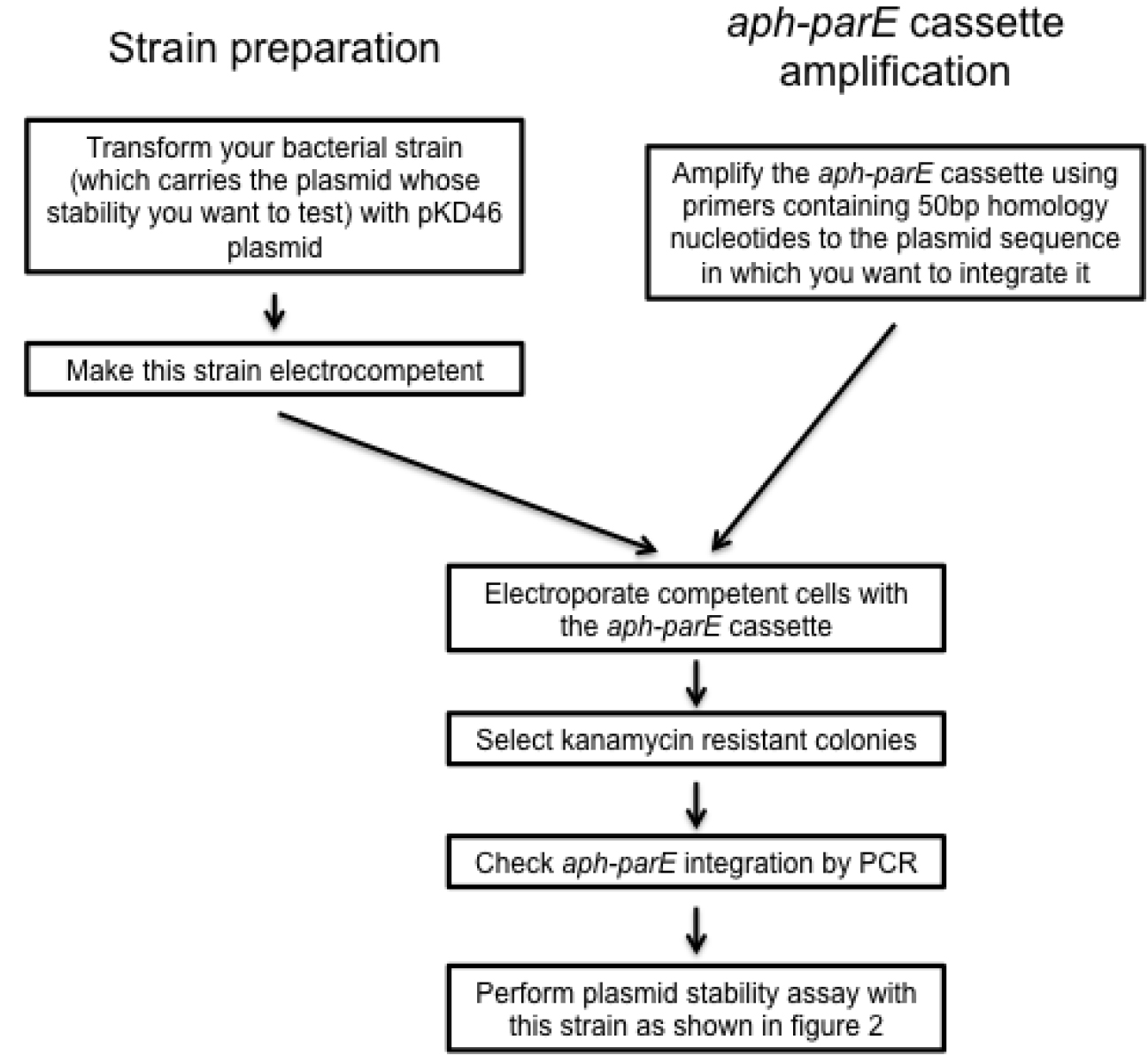
Figure 3. Scheme summarizing the design of recombinant plasmid variants
- Amplify aph-parE cassette by polymerase chain reaction (PCR) using pKD267 plasmid as template and Expand High Fidelity DNA polymerase (~1,800 bp) (see Notes 1 and 2, Table 1 and Figure 1). It is recommended to use the following PCR program: 94 °C 5 min, [94 °C 30 sec; 60 °C 30 sec; 72 °C 2 min] x 10, [94 °C 30 sec; 60 °C 30 sec; 72 °C 2 min + 5 sec increment per cycle] x 25 cycles.
- Plasmid stability assay
Note: See Note 6 before starting the assay.- Inoculate the bacterial culture in 10 ml of LB without selective pressure in a 100 ml flask (10:1 flask:medium volume ratio) at 37 °C and 150 rpm. Always adjust the amount of inoculum of all used strains by measuring the optical density (600 nm). Grow cultures according to the number of desired bacterial generations (see Notes 7 and 8). For instance, this protocol was optimized for Salmonella enterica subsp. enterica serovar Typhimurium grown for 16 h at 37 °C (~10 generations).
- Collect 1 ml of grown cultures in 1.5 ml Eppendorf tubes.
- Centrifuge for 1 min at ~10,800 x g at room temperature using a benchtop centrifuge (MiniSpin® Eppendorf), or an equivalent speed in a different benchtop centrifuge.
- Discard supernatants and wash bacterial pellets twice with 1 ml of phosphate buffered saline (PBS) pH 7.4 (see Note 9). Resuspend bacterial pellets in 1 ml PBS.
- Perform 1:10 serial dilutions using 100 µl of PBS-resuspended bacterial cultures in PBS-containing Eppendorf tubes.
- Plate 100 µl of the appropriate dilutions onto LB-plates and M9-rhamnose plates by using glass beads. Proper dilution must be adjusted according to the studied plasmid (see Notes 10 and 11).
- Incubate plates for 24 h (LB-agar) or 48-72 h (M9-rhamnose-agar) at 37 °C before counting the number of colony forming units (CFUs) (see Note 12).
- To discard false positives, CFUs grown in M9-rhamnose-agar should be tested for their kanamycin resistance by streaking them onto antibiotic-containing LB plates.
- Inoculate the bacterial culture in 10 ml of LB without selective pressure in a 100 ml flask (10:1 flask:medium volume ratio) at 37 °C and 150 rpm. Always adjust the amount of inoculum of all used strains by measuring the optical density (600 nm). Grow cultures according to the number of desired bacterial generations (see Notes 7 and 8). For instance, this protocol was optimized for Salmonella enterica subsp. enterica serovar Typhimurium grown for 16 h at 37 °C (~10 generations).
Data analysis
- Count the number of CFUs in M9-rhamnose and in LB plates
- The segregants fraction (the fraction of bacteria that have lost the studied plasmid) can be calculated as the ‘number CFUs per ml in M9-rhamnose’/‘number CFUs per ml in LB’ (Lobato-Marquez et al., 2016).
- Analyze stability differences between strains by t-test or one-way ANOVA. If ANOVA is used post hoc tests can be employed to determine mean differences between all strains (Tukey’s test) or to compare test strains to the control strain (Dunnett’s test). We suggest using GraphPad Prism software (La Jolla, USA)
Notes
- This stability assay is based on the insertion of the aph-parE cassette in the plasmid/gene of interest. The cassette integration procedure is adapted from the original method described by Datsenko and Wanner (2000) and summarized in Figure 1.
- Primer design and aph-parE amplification: design oligonucleotides that bind to the aph-parE region of pKD267 plasmid (Table 1). Primers must contain 50 bp homologous to the upstream (forward) and downstream (reverse) regions of the desired insertion site, in the 5’-end of the sequence annealing with pKD267 (Figure 1, Table 1). To test the stability of the plasmid of interest, the aph-parE cassette must be inserted into a region of the plasmid which is not involved in plasmid stability. If testing the contribution of a specific gene to plasmid stability, the aph-parE cassette must be inserted in such a way that the gene of interest is disrupted (preferentially also inactivating the promoter of that gene, to avoid polar effects).
Table 1. DNA sequence of the 3’-end oligonucleotides annealing with the pKD267 aph-parE cassettePrimer name Primer sequence (5’-3’) Used for Forward 3’-end TCTCTACGCCGGACGCATCGTG Amplify aph-parE cassette Reverse 3’-end ACTGATCAGTGATAAGCTGTC Amplify aph-parE cassette Km-Comp5 CACGATGCGTCCGGCGTAGAG Check aph-parE insertions Km-Comp3 GACAGCTTATCACTGATCAG Check aph-parE insertions
When designing the forward 3’-end and reverse 3’-end primers, it is necessary to add the 50 bp homologous to the insertion site (see Figure 1). - When purifying the aph-parE cassette it is important to remove as much of the salts as possible to prevent interferences with subsequent electroporation.
- The pKD46 plasmid is a thermo-sensitive plasmid encoding λ-Red recombinase necessary to integrate the aph-parE cassette into the desired region of the plasmid. λ-Red recombinase synthesis is induced in the presence of arabinose. Once transformed with pKD46 plasmid, the resulting recombinant strain must be grown at 30 °C and in the presence of 50 µg/ml of ampicillin until electroporation with the aph-parE fragment.
- After insertion of aph-parE cassette into the plasmid of interest it is highly recommended to always grow cells in the presence of 0.2% glucose. Glucose represses the PparE promoter and so avoids any possible toxicity derived from ParE synthesis.
- Stability assay: always grow bacteria in the presence of 50 µg/ml of kanamycin before counting the number of desired generations. This will ensure starting the experiment begins with ~100% bacteria-containing plasmid.
- If several strains containing different plasmid derivatives are being compared, the researcher should confirm that the growth rate of all strains is the same. Different growth rates mean a different number of bacterial generations and would therefore produce differences in the fraction of segregants.
- This methodology was used to measure plasmid stability of pSLT plasmid after ~10 generations and was able to detect ~1 segregant in 2 x 106 bacteria (Lobato-Marquez et al., 2016). Due to the high sensitivity of the described assay we strongly believe that this procedure might be adapted to identify plasmid-free cells in a fewer number of generations.
- After growing bacterial cultures under selected conditions, culture aliquots must be washed twice with 1 ml of PBS to remove traces of LB medium. M9-rhamnose-agar plates must contain rhamnose as the only carbon source. The rhamnose promoter is subjected to catabolic repression, meaning that other carbon sources such as glucose repress the expression of parE and therefore prevent the negative selection of plasmid-free cells.
- PBS, LB medium and M9-rhamnose plates must be kept sterile. If bacterial cultures containing aph-parE cassette are contaminated with a non aph-parE-containing strain it will produce false positive colonies in M9-rhamnose plates also sensitive to kanamycin.
- Working dilutions: as an example, for the highly stable pSLT virulence plasmid of S. typhimurium (~10-7 segregants per cell generation), 1:107 dilution was used to quantify total bacterial population in LB-agar plates, and dilutions in the range 1:1-103 were used to determine the number of segregants in M9-rhamnose-agar plates.
- M9-rhamnose plates should not be kept for more than 72 h at 37 °C, except for those bacteria with an extremely low duplication rate. We have observed that ParE toxin activity is more bacteriostatic than bactericidal, meaning that after a long time at 37 °C incubation plasmid-containing cells will grow.
Recipes
- Luria Bertani (LB) broth
10 g/L Bacto tryptone
5 g/L Bacto yeast extract
10 g/L NaCl
Sterilize by autoclaving at 120 °C for 20 min - Luria Bertani plates
LB medium
15 g/L agar
Sterilize by autoclaving at 120 °C for 20 min - M9 (10x)
176.5 g/L Na2HPO4
30 g/L KH2PO4
5 g/L NaCl
10 g/L NH4Cl
Filter sterilize - CaCl2/MgSO4 solution (100x)
0.01 M CaCl2
0.1 M MgSO4
Sterilize by filtering using a 0.22 µm pore Millipore filter or autoclaving at 120 °C for 20 min - M9 minimal medium
1x M9
1x CaCl2/MgSO4 solution
1 mg/ml Vitamine B1 (thiamine)
0.5% rhamnose
Sterilize water by autoclaving and add the rest of the sterile components. Do not autoclave the final solution. This solution can be kept at room temperature but should be kept protected from light to preserve thiamine integrity - M9-rhamnose plates
M9 minimal medium
15 g/L agar
Sterilize water containing agar by autoclaving at 120 °C for 20 min, and add the rest of the sterile components - Phosphate saline buffered (PBS)
8 g/L NaCl
0.2 g/L KCl
2.89 g/L Na2HPO4·12H2O
0.2 g/L KH2PO4
Adjust pH to 7.4 - 10% sterile glycerol
Dilute glycerol in MilliQ water to a final concentration of 10% (v/v)
Sterilize by autoclaving at 120 °C for 20 min - L-rhamnose
To prepare a sterile rhamnose solution (we recommend preparing a 15% stock solution), dilute rhamnose in MilliQ water and filter using a 0.22 µm pore Millipore filter
Acknowledgments
This work was performed in the laboratories of Prof. Díaz-Orejas and Prof. García-del Portillo and supported by grants BFU2011-25939, CSD2008-00013, and BIO2013-46281-P/BIO2015-69085-REDC from the Spanish Ministry of Economy and Competitiveness.
We thank Alexandra Willis for her critical review of the manuscript.
This protocol was originally described in Lobato-Márquez et al., 2016.
References
- Bochner, B. R., Huang, H. C., Schieven, G. L. and Ames, B. N. (1980). Positive selection for loss of tetracycline resistance. J Bacteriol 143(2): 926-933.
- Datsenko, K. A. and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97(12): 6640-6645.
- del Solar, G. H., Puyet, A. and Espinosa, M. (1987). Initiation signals for the conversion of single stranded to double stranded DNA forms in the streptococcal plasmid pLS1. Nucleic Acids Res 15(14): 5561-5580.
- Garcia-Quintanilla, M., Prieto, A. I., Barnes, L., Ramos-Morales, F. and Casadesus, J. (2006). Bile-induced curing of the virulence plasmid in Salmonella enterica serovar Typhimurium. J Bacteriol 188(22): 7963-7965.
- Gerdes, K., Larsen, J. E. and Molin, S. (1985). Stable inheritance of plasmid R1 requires two different loci. J Bacteriol 161(1): 292-298.
- Jiang, Y., Pogliano, J., Helinski, D. R. and Konieczny, I. (2002). ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol Microbiol 44(4): 971-979.
- Li, X. T., Thomason, L. C., Sawitzke, J. A., Costantino, N. and Court, D. L. (2013). Positive and negative selection using the tetA-sacB cassette: recombineering and P1 transduction in Escherichia coli. Nucleic Acids Res 41(22): e204.
- Lobato-Marquez, D., Molina-Garcia, L., Moreno-Cordoba, I., Garcia-Del Portillo, F. and Diaz-Orejas, R. (2016). Stabilization of the virulence plasmid pSLT of Salmonella Typhimurium by three maintenance systems and its evaluation by using a new stability test. Front Mol Biosci 3: 66.
- Maisonneuve, E., Shakespeare, L. J., Jorgensen, M. G. and Gerdes, K. (2011). Bacterial persistence by RNA endonucleases. Proc Natl Acad Sci U S A 108(32): 13206-13211.
- Maloy, S. R. and Nunn, W. D. (1981). Selection for loss of tetracycline resistance by Escherichia coli. J Bacteriol 145(2): 1110-1111.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Lobato-Márquez, D. and Molina-García, L. (2017). Evaluation of Plasmid Stability by Negative Selection in Gram-negative Bacteria. Bio-protocol 7(9): e2261. DOI: 10.21769/BioProtoc.2261.
Category
Microbiology > Microbial genetics > Plasmid
Molecular Biology > DNA > Extrachromosomal DNA
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.