- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Physical Removal of the Midbody Remnant from Polarised Epithelial Cells Using Take-Up by Suction Pressure (TUSP)

Published: Vol 7, Iss 8, Apr 20, 2017 DOI: 10.21769/BioProtoc.2244 Views: 8992
Reviewed by: Andrea PuharJingli CaoThirupugal Govindarajan
Original research article
The authors used this protocol in:

Aug 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
In polarised epithelial cells the midbody forms at the apical cell surface during cytokinesis. Once severed, the midbody is inherited by one of the daughter cells remaining tethered to the apical plasma membrane where it participates in non-cytokinetic processes, such as primary ciliogenesis. Here, we describe a novel method to physically remove the midbody remnant from cells and assess the possible effects caused by its loss (Bernabé-Rubio et al., 2016).
Keywords: Epithelial cellsBackground
The midbody or the Flemming body is the central part of the intercellular bridge formed between daughter cells during the final stages of mitosis. The abscission on either side of the bridge by the endosomal sorting complexes required for transport (ESCRT) machinery, results in the physical separation of the two daughter cells (Green et al., 2012). In addition to its known function in the regulation of mitosis, recent studies have begun to elucidate post-mitotic roles for the midbody. Due to its role in the initiation of lumen formation in kidney cells, the midbody has been postulated to serve as a polarity cue (Li et al., 2014). More recently, it has been demonstrated that the midbody remnant is directly involved in primary ciliogenesis by polarised Madin-Darby canine kidney (MDCK) cells (Bernabé-Rubio et al., 2016). It has been also found to have a role in formation of the dorsoventral axis during the development of Caenorhabditis elegans (Singh and Pohl, 2014), and in defining cell fate and differentiation (Kuo et al., 2011). Previous studies have used laser ablation to impair the function of the midbody remnant. When performed in cultured cell lines, however, laser ablation can result in cell death due to damage of the plasma membrane and proximal cytosolic elements. Accordingly, we have designed a gentle procedure, which we have called ‘take-up by suction pressure’ (TUSP). TUSP allows non-deleterious midbody remnant removal from the cell surface of epithelial cells. The fundamental principle is based on using a fine-aperture glass pipette attached to patch-clamp apparatus to physically remove the midbody with applied negative pressure (Figure 1).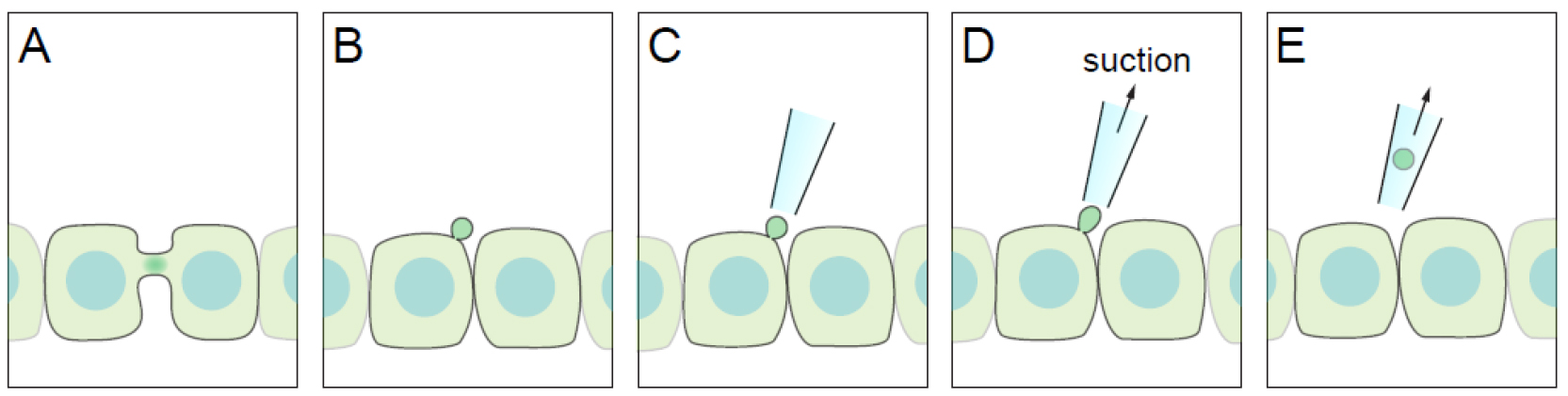
Figure 1. Diagram of the TUSP procedure. A. An apical intercellular bridge forms during cytokinesis in polarised epithelial cells. B. After abscission, one of the daughter cells inherits the midbody as a remnant, which will be positioned over the apical cell surface. C-E. By using a glass pipette connected to path-clamp apparatus, the midbody remnant can be removed from cells if suction pressure is applied.
Materials and Reagents
- 12 mm glass coverslips #1 (VWR, catalog number: 631-0713 )
- Gridded coverslips (optional) (Electron Microscopy Sciences, catalog number: 72265-12 )
- Falcon 24-well plates (Corning, catalog number: 353047 )
- Permanent marker (Faber-Castell Multimark 1523) (CultPens, catalog number: FC19628 )
- 1 mL syringe (BD, catalog number: 303172 )
- 25 G 1 ½ needle (BD, catalog number: 305127 )
- Epithelial Madin-Darby canine kidney (MDCK II) from ATCC (ATCC, catalog number: CRL2936 )
- DNA construct expressing a fluorescent midbody localised protein (e.g., Cherry-tubulin, Addgene, catalog number: 49149 )
- Dulbecco’s modified Eagle’s medium (DMEM) (Sigma-Aldrich, catalog number: D5796 )
- Fetal bovine serum (Sigma-Aldrich, catalog number: F7524 )
- Penicillin-streptomycin solution (Sigma-Aldrich, catalog number: P4333 )
- Lipofectamine 2000 (Thermo Fisher Scientific, InvitrogenTM, catalog number: 11668027 )
- Hank’s balanced salt solution (HBSS) without phenol red (Sigma-Aldrich, catalog number: H8264 )
- 1 M HEPES solution (Thermo Fisher Scientific, GibcoTM, catalog number: 15630080 )
Equipment
- Autoclave
- Tweezers (Fine Science Tools, catalog number: 11251-20 )
- LSM 710 confocal microscope (ZEISS, model: LSM 710 ) or any other inverted confocal microscope with 25x and 40x oil objectives and a numerical aperture of 0.8 and 1.3, respectively
- Patch clamp equipment (Axon Instruments)
- Microscope BX51 (Olympus, model: BX51 )
- Borosilicate glass with filament for pipette fabrication. Outer diameter: 1.5 mm, inner diameter: 0.86 mm, 10 cm length (Linton Instrumentation, catalog number: BF150-86-10 )
- CO2 cell culture incubator (Thermo Electron Corporation)
- P-97 Flaming/Brown micropipette puller (Sutter Instruments, model: P-97 )
- Sutter MP-225 motorised micromanipulators (Sutter Instruments, model: MP-225 )
Software
- ImageJ (https://imagej.nih.gov/ij, National Institutes of Health)
Procedure
- Using permanent marker, label a round coverslip with a small circle and one extra mark to keep the same orientation of the coverslip when moving between different equipment (Figure 2). Autoclave the coverslips to avoid contaminations.
Note: Instead of using coverslips labelled with permanent marker, gridded coverslips can be used for subconfluent cell cultures. In confluent fully polarised monolayers, however, the grids can be hard to visualise.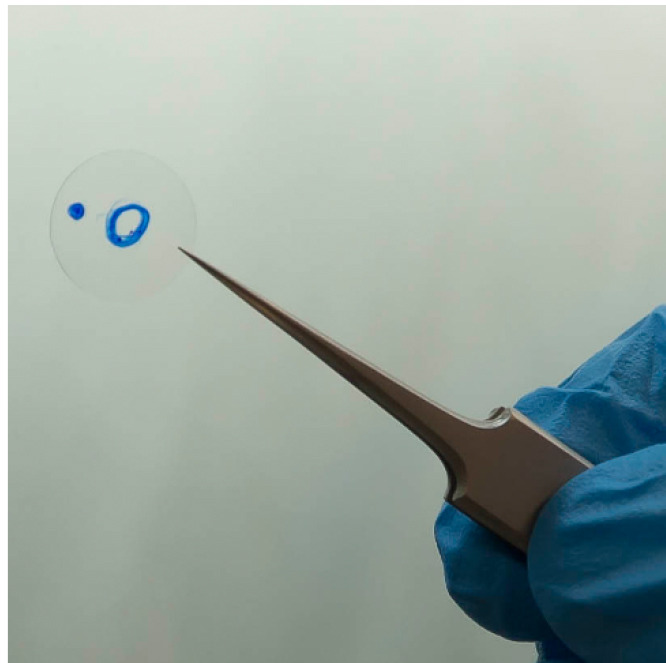
Figure 2. Round coverslip labelled with permanent marks for generation of cell maps - Place the coverslip into a 24-well plate and seed 200,000 MDCK cells onto the coverslip, which should be oriented so that the unmarked side is on top. Grow the cells in DMEM supplemented with 5% fetal bovine serum, 50 U/ml penicillin, and 50 µg/ml streptomycin.
- The following day transfect MDCK cells with a midbody localised protein (e.g., PRC1, MKLP1, or tubulin) genetically fused to a fluorescent protein such as GFP or mCherry. If using stably transfected cells, between 20-60% should be expressing the transgene. Cells can be diluted with untransfected cells to achieve this. It is important to have between 20-60% of cells expressing the transgene in order to have a distinctive cell map allowing easy orientation of the cell map at the later stages. Leave cells to grow for at least two days to allow cells to divide and subsequently generate new midbodies.
Note: We recommend using Lipofectamine 2000 reagent according to the manufacturers’ instructions for transfection of MDCK cells. In this protocol we use 200,000 cells, 1 µg of DNA, and 1 µl of Lipofectamine 2000 per well. - For the cell map, image the cells situated inside the circle using a confocal microscope at low magnification (Figure 3). Print out the images allowing them to be used for reference in the later steps.
Note: For the generation of cell maps, we use a 25x objective.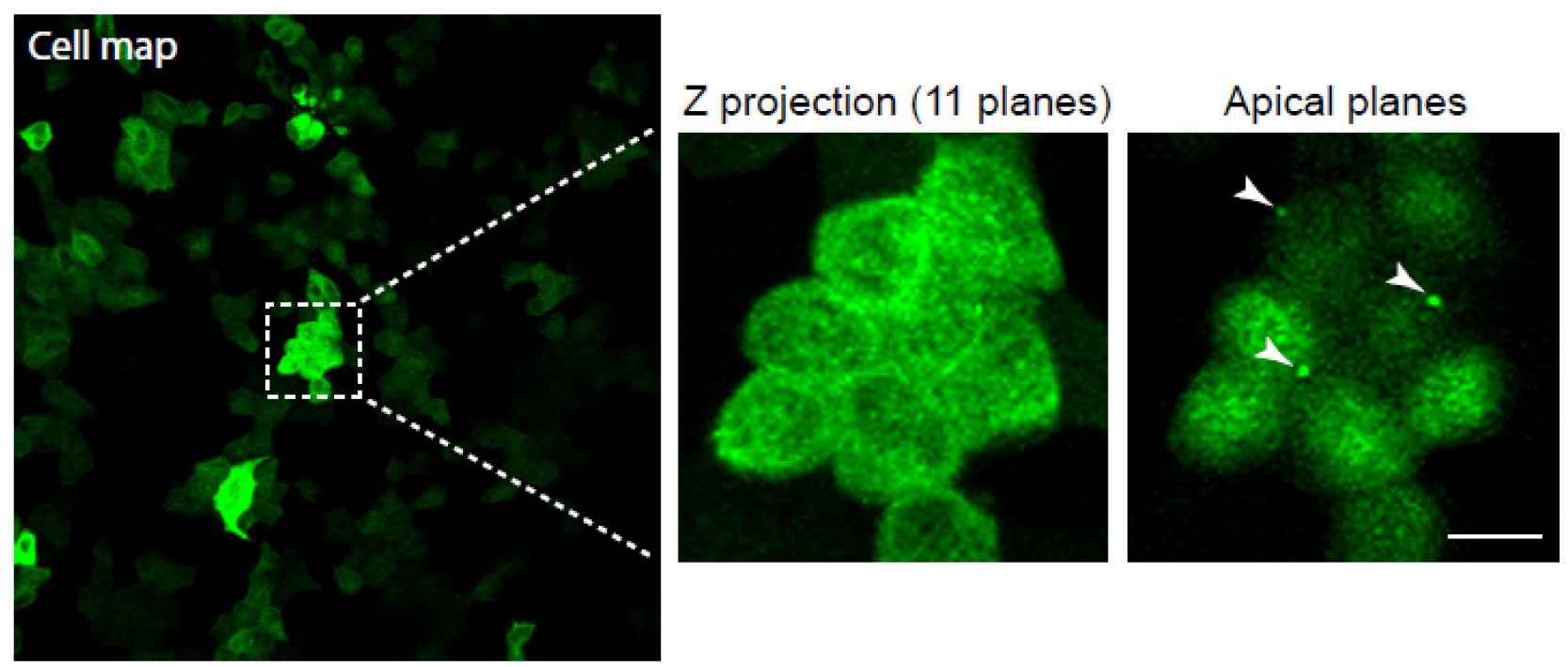
Figure 3. Generation of a cell map and midbody visualisation. Cells stably expressing GFP-tubulin were used. Cell map is represented on the far left using a 25x objective. The boxed region shows a magnification with a 40x objective, and was used for localising cells exhibiting midbodies. Note that the midbodies are localised on the apical surface. The white arrowheads mark the midbody remnants. Scale bar = 10 µm. - Subsequently, choose some cells to image at higher resolution to document the presence of the midbody remnants before manipulation with patch-clamp apparatus. Cells that are amenable to midbody remnant removal will have the remnants positioned over the apical cell surface (Figure 3; Video 1). Using a pen, make a note of the cells imaged on the printed image.
Note: Be careful to avoid photobleaching. Move focal plane to the apical surface of the cell monolayer to find midbody remnants.
 Video 1. Z-stack images of MDCK cells stably expressing GFP-tubulin before TUSP. Images are shown from the basolateral to apical membranes (i.e., bottom to the top). The white arrowheads point to the midbody remnants. Note that the remnants are localised at the apical cell surface.
Video 1. Z-stack images of MDCK cells stably expressing GFP-tubulin before TUSP. Images are shown from the basolateral to apical membranes (i.e., bottom to the top). The white arrowheads point to the midbody remnants. Note that the remnants are localised at the apical cell surface. - Transfer samples to the patch-clamp apparatus (Figure 4). Fill the chamber of the patch-clamp apparatus with HBSS medium supplemented with 0.5% fetal bovine serum, HEPES 20 mM, pH 7.2-7.5, 50 U/ml penicillin, and 50 µg/ml streptomycin, and place the coverslip into the chamber. To allow easier detection of previously imaged areas, use the marked position on the coverslip to position the sample in the same orientation as was previously used to generate the cell map.

Figure 4. Patch-clamp apparatus coupled to an epifluorescence microscope - Using the printed cell maps as a reference, identify the cells previously imaged inside the circle under the epifluorescence microscope coupled to the patch-clamp apparatus.
Note: In this step, use a low magnification objective. - Once the cell map has been correctly oriented, use an objective of higher magnification and move along the XY axis to the position where the cells whose midbody remnants were documented before, are situated. Double-check for the presence of remnants.
Note: In our epifluorescence microscope this step is performed with a 60x objective. Take a quick look to observe the remnants and turn off the fluorescence channel to avoid photobleaching. - Move the objective to allow the glass pipette to be between the objective and the monolayer.
- For fabricating a glass pipette, load a borosilicate glass pipette of 1.5 mm in outer diameter, 0.86 mm in inner diameter, and 10 cm in length into a micropipette puller (Figure 5). Ramp value = 515-535; Pull = 0; Velocity = 15-25; 4 heating cycles.

Figure 5. Puller used for creating glass pipettes - Once generated, take the glass pipette, and fill it with HBSS medium using a syringe with a 25 G 1 ½ needle.
- Fit the glass pipette to the pipette holder, and move it under the objective (Figure 6).

Figure 6. Glass pipette fitted to the pipette holder of the patch-clamp apparatus should be positioned in between the objective and the sample. The red circle indicates the glass pipette. - Use brightfield illumination to focus on the tip of the glass pipette.
- Simultaneously move down the pipette and the objective of the microscope on the z axis until the monolayer comes into the focal plane. Do not let the glass pipette contact the monolayer at this stage.
- Once the monolayer is in focus, use fluorescence to detect the midbody remnants (Figure 7; Video 2; Bernabé-Rubio et al., 2016).
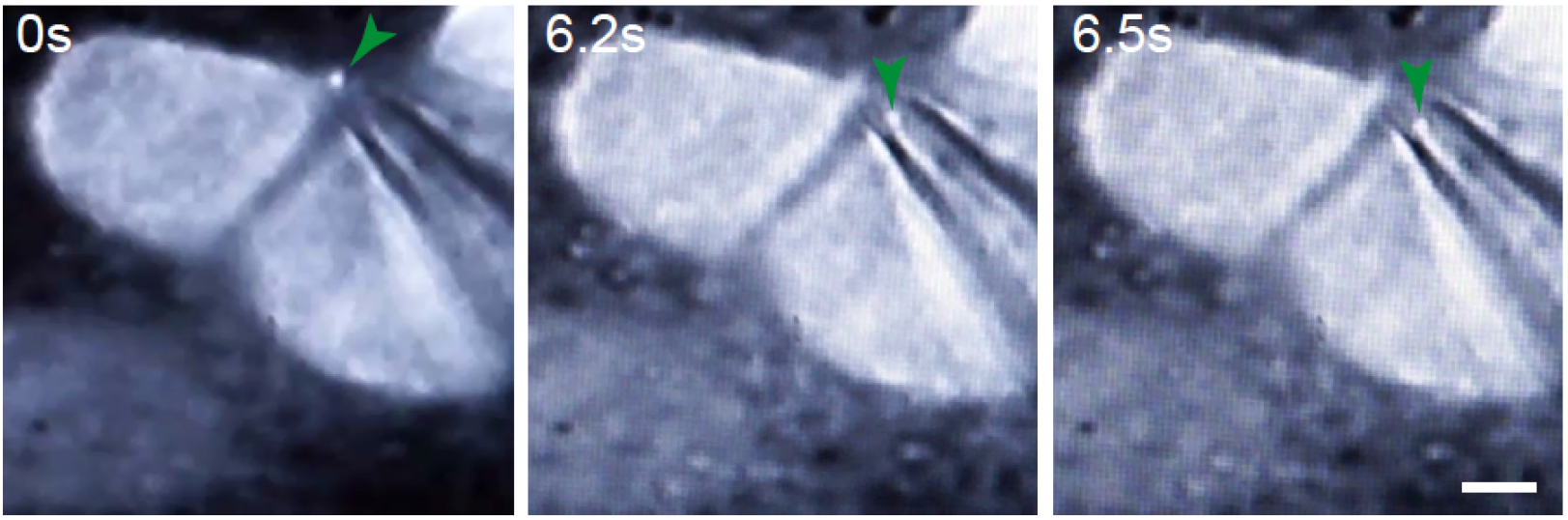
Figure 7. Representative example of cells subjected to TUSP. MDCK cells expressing GFP-tubulin were imaged at real time under an epifluorescence microscope. Green arrows indicate the midbody remnant. Scale bar = 5 μm.
Video 2. Videomicroscopy of the TUSP procedure. MDCK cells stably expressing GFP-tubulin were subjected to TUSP. Images were captured in real time. Brightfield and fluorescence channels are shown superimposed. The green arrow indicates the position of the midbody remnant. - Select a midbody remnant for removal and slowly move the glass pipette closer to the remnant using the brightfield illumination. Once proximal to each other, keep the brightfield channel and the fluorescence superimposed. Using the mouth to apply negative pressure to the pipette via a tube, draw the midbody remnant from the cell surface (Figures 1 and 7; Video 2; Bernabé-Rubio et al., 2016).
- After midbody remnant removal, image the cells under a confocal microscope acquiring different planes of the cell to ensure that removal of the whole midbody has occurred (Figure 8; Video 3).
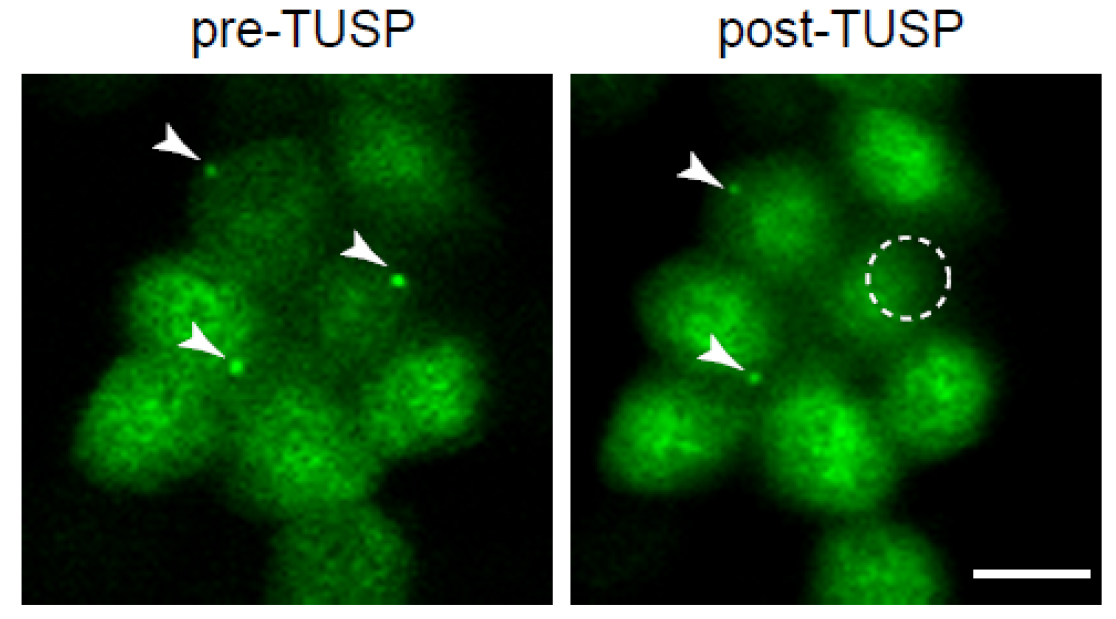
Figure 8. Midbody remnants from cells expressing GFP-tubulin were removed by TUSP. Cells exhibiting midbodies are shown before (pre-TUSP) and after TUSP (post-TUSP). Note that the remnants are localised at the apical surface. The white arrowheads mark the midbody remnants. Since cells continuously move, it is often difficult to have several cells in-focus while imaging a cell field. The circle indicates the zone subjected to TUSP. Note that the midbody remnant of the cell exposed to TUSP was removed. Scale bar = 10 µm.
Video 3. Z-stack images of MDCK cells stably expressing GFP-tubulin before (pre-TUSP) and after TUSP (post-TUSP). Images are shown from the basolateral to apical membranes (i.e., bottom to the top). The white arrowheads point to the midbody remnants. The circle indicates the zone subjected to TUSP. Note that the remnant of the cell subjected to TUSP was completely removed. - Place cells in an incubator at 37 °C in DMEM supplemented with 5% fetal bovine serum, 50 U/ml penicillin, and 50 µg/ml streptomycin under an atmosphere of 5% CO2/95% air.
- Various readouts can be used to assess possible biological effects of midbody removal using appropriate experimental setups. For example, we have studied the role of the midbody remnant in primary ciliogenesis using fluorescence microscopy (Bernabé-Rubio et al., 2016).
Data analysis
For qualitative analysis of effects of midbody removal live-cell videomicroscopy can be used. This procedure can also be used to quantitatively address the absence or presence of the cilium 24 h after midbody removal, as used previously (Bernabé-Rubio et al., 2016). Cells should be fixed after 24 h and imaged to detect the presence of the cilium. In this case the data is categorical and as such a chi-squared analysis is appropriate after a minimum of 3 experimental repetitions with controls.
Notes
This protocol is highly reproducible as long as there is no phototoxicity/photobleaching of cells, and cell maps are clearly defined. A short exposure time to light is recommended to reduce phototoxicity/photobleaching.
Recipes
- HBSS medium to fill the chamber of the patch-clamp apparatus
Hank’s balanced salt solution (HBSS) without phenol red
20 mM HEPEs (pH 7.2-7.5)
50 U/ml penicillin, 50 µg/ml streptomycin
0.5% fetal bovine serum - DMEM for MDCK cell culture
Dulbecco’s modified Eagle’s medium (DMEM)
50 U/ml penicillin, 50 µg/ml streptomycin
5% fetal bovine serum
Acknowledgments
We thank Dr. José A. Esteban (Centro de Biología Molecular Severo Ochoa, Madrid, Spain) for the use of his patch-clamp equipment. We express our gratitude to Laura Rangel for taking some of the pictures shown in this protocol, and for recording the video of TUSP procedure. We also thank Minerva Bosch-Fortea for critical reading of the protocol. D.C.G. would like to thank Juan Bonifacino and the Bonifacino Lab for support whilst developing this protocol, and Laura Pellegrini for critically reading the protocol. The expert technical advice of the Optical and Confocal Microscopy Unit of the Centro de Biología Molecular Severo Ochoa is gratefully acknowledged. This work was supported by grant BFU2015-67266-R from the Spanish Ministerio de Economía y Competitividad/Fondo Europeo de Desarrollo Regional (MINECO/FEDER) to M.A. Alonso. M. Bernabé-Rubio is the holder of a fellowship from the Ministerio de Economía y Competitividad.
References
- Bernabé-Rubio, M., Andres, G., Casares-Arias, J., Fernandez-Barrera, J., Rangel, L., Reglero-Real, N., Gershlick, D. C., Fernandez, J. J., Millan, J., Correas, I., Miguez, D. G. and Alonso, M. A. (2016). Novel role for the midbody in primary ciliogenesis by polarized epithelial cells. J Cell Biol 214(3): 259-273.
- Green, R. A., Paluch, E. and Oegema, K. (2012). Cytokinesis in animal cells. Annu Rev Cell Dev Biol 28: 29-58.
- Kuo, T. C., Chen, C. T., Baron, D., Onder, T. T., Loewer, S., Almeida, S., Weismann, C. M., Xu, P., Houghton, J. M. and Gao, F. B., Daley, G. Q. and Doxsey, S. (2011). Midbody accumulation through evasion of autophagy contributes to cellular reprogramming and tumorigenicity. Nat Cell Biol 13: 1214-1223.
- Li, D., Mangan, A., Cicchini, L., Margolis, B. and Prekeris, R. (2014). FIP5 phosphorylation during mitosis regulates apical trafficking and lumenogenesis. EMBO Rep 15(4): 428-437.
- Singh, D. and Pohl, C. (2014). Coupling of rotational cortical flow, asymmetric midbody positioning, and spindle rotation mediates dorsoventral axis formation in C. elegans. Dev Cell 28(3): 253-267.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Bernabé-Rubio, M., Gershlick, D. C. and Alonso, M. A. (2017). Physical Removal of the Midbody Remnant from Polarised Epithelial Cells Using Take-Up by Suction Pressure (TUSP). Bio-protocol 7(8): e2244. DOI: 10.21769/BioProtoc.2244.
- Bernabé-Rubio, M., Andres, G., Casares-Arias, J., Fernandez-Barrera, J., Rangel, L., Reglero-Real, N., Gershlick, D. C., Fernandez, J. J., Millan, J., Correas, I., Miguez, D. G. and Alonso, M. A. (2016). Novel roce for the midbody in primary ciliogensis by polarized epithelial cells.J Cell Biol 214(3): 259-273.
Category
Developmental Biology > Morphogenesis > Cell structure
Cell Biology > Organelle isolation > Midbody
Cell Biology > Cell structure > Cell surface
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.