- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Measurement of FNR-NrdI Interaction by Microscale Thermophoresis (MST)
Published: Vol 7, Iss 8, Apr 20, 2017 DOI: 10.21769/BioProtoc.2223 Views: 12455
Reviewed by: Yanjie LiAndré Alex GrassmannAnonymous reviewer(s)
Original research article
The authors used this protocol in:
Sep 2016
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
This protocol describes how to measure protein-protein interactions by microscale thermophoresis (MST) using the MonolithTM NT.115 instrument (NanoTemper). We have used the protocol to determine the binding affinities between three different flavodoxin reductases (FNRs) and a flavodoxin-like protein, NrdI, from Bacillus cereus (Lofstad et al., 2016). NrdI is essential in the activation of the manganese-bound form of the class Ib ribonucleotide reductase (RNR) system. RNRs, in turn, are the only source of the de novo synthesis of deoxyribonucleotides required for DNA replication and repair in all living organisms.
Keywords: MSTBackground
Protein-protein interactions are often characterised in terms of the associated dissociation constant, KD. The binding constant can be established using a variety of techniques, such as isothermal calorimetry (ITC), NMR spectroscopy, and surface plasmon resonance (SPR). An alternative method is based on thermophoresis, a phenomenon where distinct molecules (such as a protein-protein complex versus individual proteins) respond differently to a temperature gradient (Duhr and Braun, 2006; Seidel et al., 2013). This method is rapid, no sample immobilisation is needed and the sample requirement is low. Briefly, one of the proteins is labelled with a fluorescent dye and kept at a constant, low concentration. A dilution series is set up, where the other protein is diluted up to 16 times, creating a vast concentration range. The two proteins are subsequently mixed and loaded into capillaries, which are scanned in the MonolithTM NT.115 instrument, developed and sold exclusively by NanoTemper. The samples are subjected to a temperature gradient, and the movement of the fluorescently labelled molecule is tracked. The difference in the fluorescence of the molecule at the initial temperature and at the new temperature is used to generate a binding curve as a function of the concentration of the unlabelled protein.
Materials and Reagents
- Tubes
15 (VWR, catalog number: 525-0400 )
50 ml (VWR, catalog number: 525-0402 ) - Pipette tips
0.1-10 μl (VWR, catalog number: 613-0735 )
1-200 μl (VWR, catalog number: 613-0740 )
100-1,250 μl (VWR, catalog number: 613-0739 ) - 0.2 μm filter (SARSTEDT, catalog number: 83.1826.001 )
- Syringe, 50 ml (BD, catalog number: 300865 )
- Protein 1 (here, NrdI, locus-tag: bc1353) and protein 2 (here, FNR1 bc0385, FNR2 bc4926, FNR3 bc1495)
- MO-L001 MonolithTM Protein Labeling Kit RED-NHS (Amine Reactive) (NanoTemper) (contains NT-647-NHS dye [store at -20 °C]; spin column for buffer exchange [store at 4 °C]; gravity flow column for purification [store at 4 °C]; labeling buffer [store at 4 °C])
- MO-K002 MonolithTM NT.115 Standard Treated Capillaries (NanoTemper) (contains MonolithTM NT.115 Standard Treated Capillaries [store at RT]; 10% Tween 20 [store at 4 °C]; Albumin fraction A [store at 4 °C]; MST buffer [store at -20 °C]; 200 μl vials)
- DMSO (Sigma-Aldrich, catalog number: D4540 )
- Liquid N2
- HEPES (AppliChem, catalog number: A3724 )
- Potassium chloride (KCl) (EMD Millipore, catalog number: 104936 )
- Tween-20
- Buffer A (see Recipes)
Equipment
- Pipettes (various sizes)
- Benchtop centrifuge
- Vortex
- MonolithTM NT.115 with Blue/Red filter (NanoTemper Technologies, model: MO-G008 )
Procedure
- Labelling the protein with fluorescent dye
- The instructions for this step are based on using a dye and consumables supplied from NanoTemper. The NanoTemper dyes (namely RED, BLUE, and GREEN) are available with either an NHS-ester group that reacts with primary amines on for example lysines, or a maleimide group that reacts with sulfhydryl groups on e.g., cysteines. Other dyes may also be used, or alternatively proteins that have a fluorescent tag, in which case no further labelling should be required. Note that your dye (or tag) of choice must be compatible with the filters that are installed in the instrument and that the dye, once bound to the protein, should not block any potential binding sites.
Note: When labelling one of your proteins with a fluorescent dye, you first need to determine which of the two proteins you want to label. It may be useful to consider that you will only use a very small amount of the labelled protein and that, depending on the KD, you might have to use much more of the unlabelled protein (at least 20x the KD). Our measured binding affinities were in the 20-50 μm range and in order to have a satisfying binding curve the protein concentrations in the capillaries were approximately 20 nM of labelled NrdI, and 1 mM FNR in capillary 1 to 15 nM in capillary 16 with a 1:1 dilution series. - We chose to label NrdI since we were testing its binding affinity to three other proteins (FNRs). NrdI was labelled with the RED-NHS dye, as it contains eight lysines and no cysteines. Based on the NrdI crystal structure (Røhr et al., 2010) we do not expect any of the lysines to be in the proximity of the binding site.
Note: We have used buffer A (see Recipes) as our labelling buffer. When using the NHS-based dyes, the protein should not be in a buffer that contains primary amines, as these might compete with the protein in the labelling reaction. Similarly, when using the maleimide-based dyes, the protein cannot be in a buffer that contains sulfhydryl groups. Also, the reducing agents DTT and β-mercaptoethanol should not be used with either dye (TCEP may be used for both dyes). If the protein is dissolved in a buffer that contains any of the above-mentioned reagents, a buffer exchange step is required prior to labelling. NanoTemper provides a labelling buffer in the labelling kit that may be used, as well as a spin column for buffer exchange.- Dilute the protein stock to a concentration that is lower than your expected KD using labelling buffer. NrdI was diluted to 25 μM and a final volume of 188 μl.
Note: NanoTemper recommends using a protein concentration between 2 and 20 μM and a volume of 100 μl. - Dissolve the dye in 100% DMSO (30 μl). Vortex well and ensure that all the dye is dissolved.
- Dilute the dye with labelling buffer to a concentration that is 2-3x the concentration of your protein. Here, the NT-647 dye was diluted to 75 μM and a final volume of 188 μl.
- Mix the protein and the dye solution in a 1:1 volume ratio and incubate at room temperature in the dark for 30 min.
Note: This is the suggested reaction condition as described in the NanoTemper labeling kit manual, but varying the dye:protein ratio or the incubation time might increase the labeling yield. - In the meantime, wash the gel filtration column that is supplied in the kit. Pour off the storage solution and equilibrate the column with 3 x 3 ml buffer A using gravity flow.
- Add the protein/dye mix to the centre of the resin in the column. When the sample has completely entered the column bed, add buffer to adjust the total volume of applied sample to 500 μl (We added 500 - (2 x 188) = 124 μl buffer). Do not collect the flow through.
- Elute the labelled protein with 600 μl buffer A and collect the flow through in a clean tube.
Note: It is also possible to collect smaller fractions (e.g., 4 x 150 μl) and check the fluorescence intensity of each fraction in order to find the fraction that contains the labelled protein. Note that free dye might elute at the end. - Perform a capillary scan (cap scan) and check the fluorescence intensity of the labelled protein.
Note: The degree of labelling should also be determined (see Notes). - Aliquot the protein in 20 μl fractions, flash freeze in liquid N2 and store at -80 °C. The fluorescently labelled NrdI protein was used within 4 months.
- Dilute the protein stock to a concentration that is lower than your expected KD using labelling buffer. NrdI was diluted to 25 μM and a final volume of 188 μl.
- The instructions for this step are based on using a dye and consumables supplied from NanoTemper. The NanoTemper dyes (namely RED, BLUE, and GREEN) are available with either an NHS-ester group that reacts with primary amines on for example lysines, or a maleimide group that reacts with sulfhydryl groups on e.g., cysteines. Other dyes may also be used, or alternatively proteins that have a fluorescent tag, in which case no further labelling should be required. Note that your dye (or tag) of choice must be compatible with the filters that are installed in the instrument and that the dye, once bound to the protein, should not block any potential binding sites.
- KD-measurement
- Spin down protein stocks at 15,000 x g for 5 min.
- Dilute the fluorescently labelled protein with buffer A to a concentration that gives a fluorescence count between 200 and 1,500 (the detection limits of the instrument). Remember that the protein will be diluted 1:1 in the actual experiment and that you might have to increase the concentration of the labelled protein to remain within the detection limit. The fluorescently labelled NrdI protein was diluted 1:25.
Note: You can adjust the counts by either changing the LED-power or the concentration of your fluorescently labelled molecule. - Perform a cap scan to ensure that the concentration is within the detection limits and that the protein is not sticking to the capillary walls (sticking is evidenced by shoulders or double peaks in the cap scan traces instead of a smooth, symmetrical curve) (Figure 1).
Note: If the protein is sticking to the capillaries, you may want to try adding detergents or additives such as Tween-20, BSA, or Pluronic F-127, changing the pH or ionic strength of your buffer, changing to a different buffer, or using the premium coated capillaries.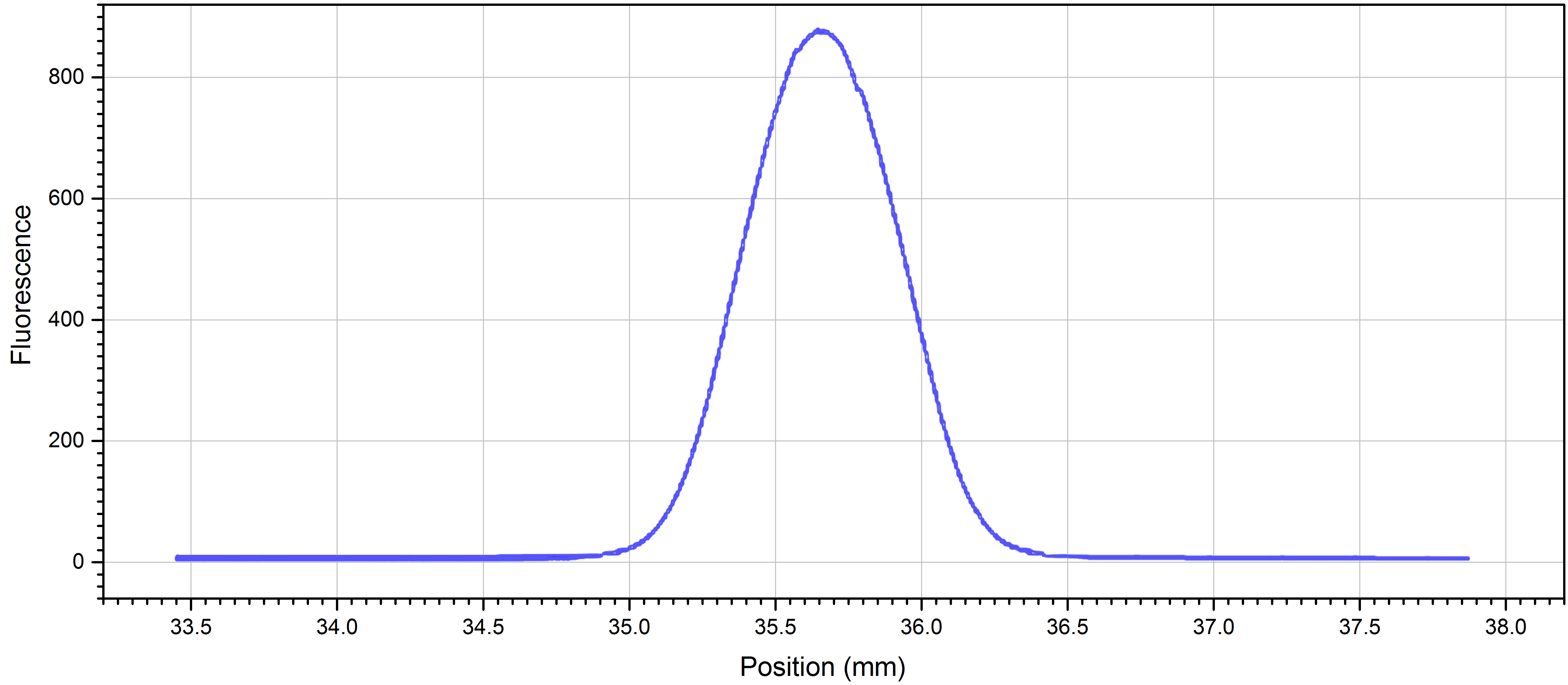
Figure 1. Cap scan of 1:25 dilution NrdI in standard capillaries at 20% LED-power - Number 16 microtubes 1-16.
Note: The tubes are provided with the capillaries. - Pipet 10 μl buffer A in tubes 2-16.
Note: Use a 10 μl pipette in steps B5, B6, and B7 to improve accuracy. - Pipet 20 μl of the unlabelled protein into tube 1. Remove 10 μl, and add these to tube 2. Mix by pipetting up and down several times. Remove 10 μl and add these to tube 3. Mix well. Continue like this for all the tubes. In tube 16, remove 10 μl and discard the liquid. You should now have 10 μl in all the tubes.
Note: The unlabelled protein must also be in the assay buffer to avoid any buffer dilution effects. You may want to use a new pipet tip for each transfer to ensure consistent volumes in the serial dilution. - Add 10 μl of the fluorescently labelled protein to all the tubes and mix well. Use a new pipet tip each time.
Note: Do not use less than 20 μl total volume of reaction mixture so that sticking and evaporation issues are minimised and pipetting errors are reduced. - Incubate the reaction mixtures for 5 min at room temperature.
- Load the samples into the capillaries and place the capillaries in the sample holder.
Note: Be careful not to touch the capillaries in the middle and avoid any bubbles in the middle of the capillary. - Put the sample holder in the MST-instrument. Perform a cap scan of all the capillaries and run the MST-experiment. The NrdI-FNR interaction was measured using 20% LED-power and 40% MST-power.
Note: The LED-power can be adjusted up to 95%, whereas the MST-power should ideally not be increased to over 40%.
- Spin down protein stocks at 15,000 x g for 5 min.
Data analysis
- Inspecting the raw data
- Evaluate the cap scans. They should be symmetrical and should not exhibit any bumps in the traces. The maximum fluorescence intensity of each peak should not differ by more than ± 10% (Figure 2).
Note: If the fluorescence differs by more than 10% in a concentration-dependent manner, it either indicates that the fluorescence is changing upon binding, or that the labelled protein is lost due to precipitation or unspecific adsorption during the sample preparation, which is carried through in the serial dilution. If the fluorescence differs by more than 10% in a non-concentration dependent manner, the assay conditions must be optimised, as described in step B3.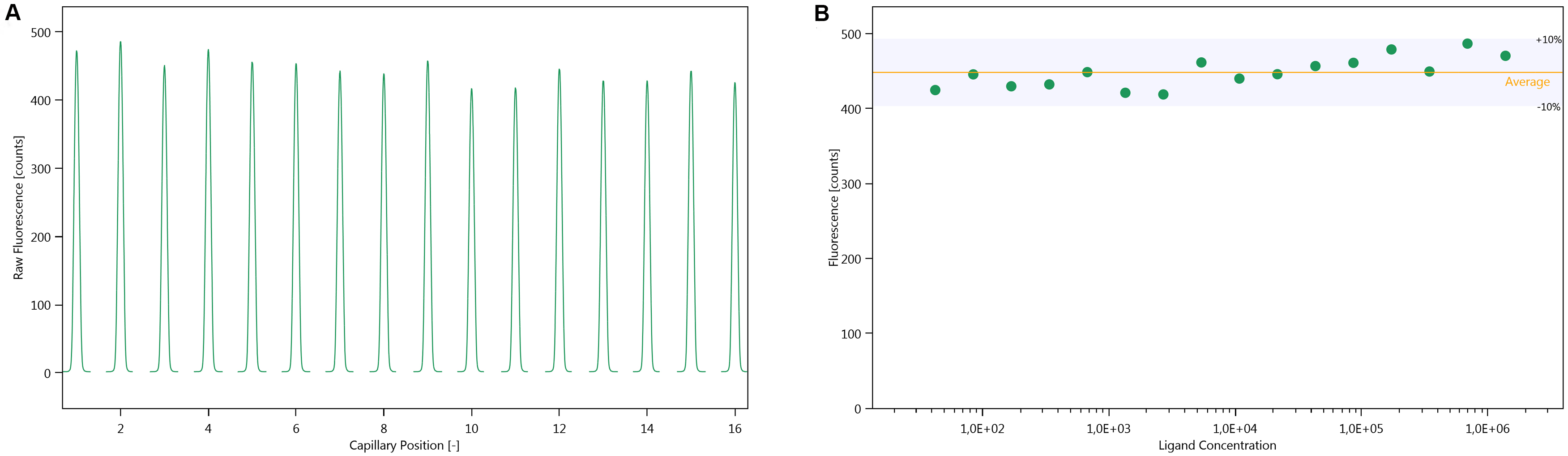
Figure 2. Cap scan inspection. A. Cap scan of a representative experiment; B. Variation in fluorescence intensity of the cap scan in A. - Inspect the MST-traces (Figure 3). Any bumps indicate that the samples are not homogenous. The traces should also not cross each other.
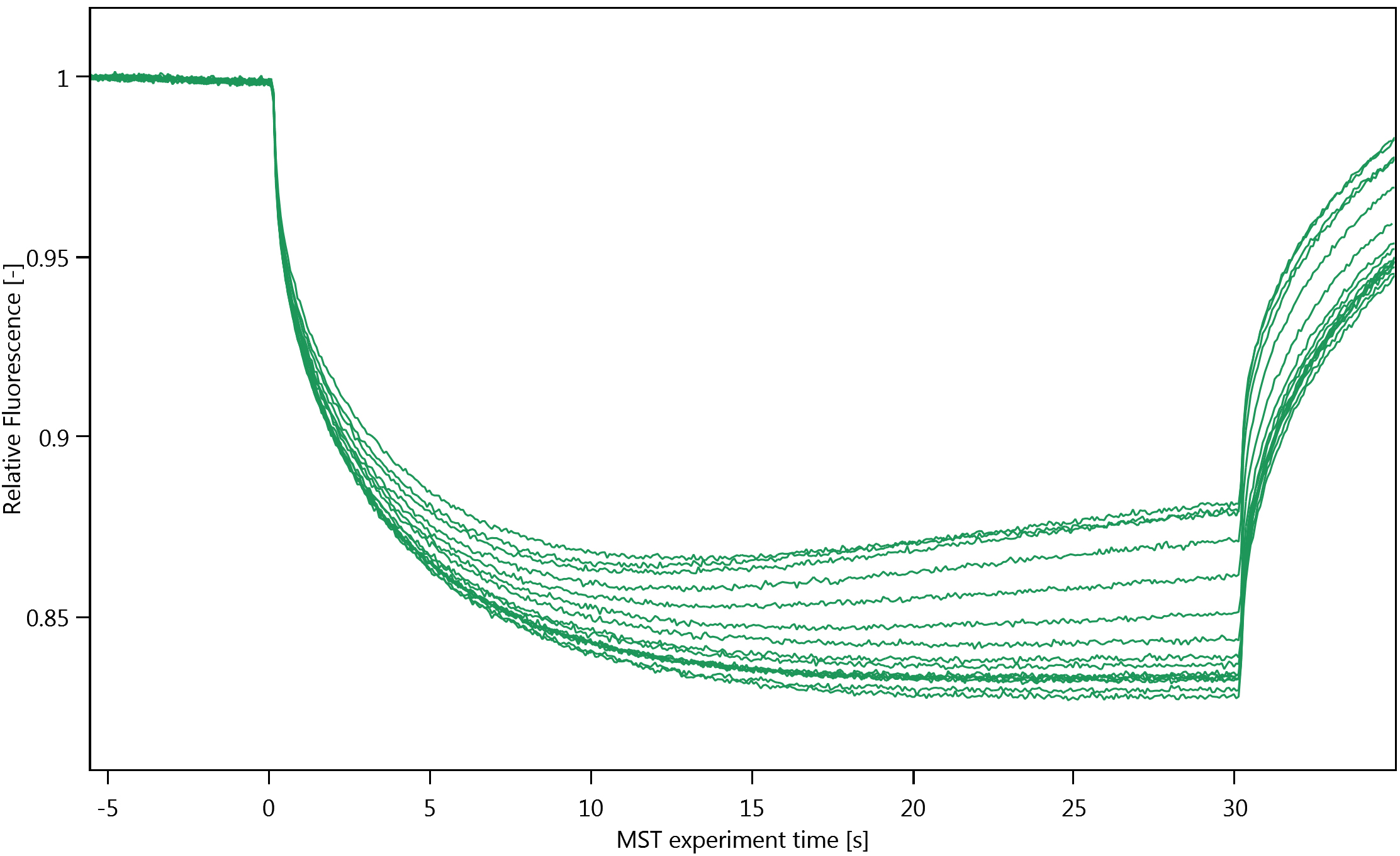 Figure 3. MST traces of a representative experimentNote: Thermophoresis can be both positive and negative. We have observed positive thermophoresis (see Figure 3), i.e., a net decrease in the observed fluorescence because the fluorescently labelled molecules move from the hotter region in the capillaries to the cooler region. We attribute the increase in the MST-traces at the end of each run to convection (Figure 3).
Figure 3. MST traces of a representative experimentNote: Thermophoresis can be both positive and negative. We have observed positive thermophoresis (see Figure 3), i.e., a net decrease in the observed fluorescence because the fluorescently labelled molecules move from the hotter region in the capillaries to the cooler region. We attribute the increase in the MST-traces at the end of each run to convection (Figure 3).
- Evaluate the cap scans. They should be symmetrical and should not exhibit any bumps in the traces. The maximum fluorescence intensity of each peak should not differ by more than ± 10% (Figure 2).
- Data processing and analysis
Data processing and analysis were carried out using the MO.Affinity Analysis program (NanoTemper). Three or four replicates were merged to form one dataset.- Select the appropriate binding model. We selected KD.
- Enter the concentration of your fluorescently labelled molecule (TargetConc) and fix this by ticking the box.
- The program will compute the KD-value and the associated uncertainty. The NrdI-FNR interaction was analysed using the ‘Thermophoresis with T jump’ evaluation strategy.
Note: The program automatically computes the binding constant taking into account both temperature jump (immediate change in fluorescence upon the temperature change) and thermophoresis effects (the movement of the molecules in the temperature gradient). This mode is called Thermophoresis with T jump. It is possible to change the evaluation strategy by selecting the ‘Expert mode’. In this mode four options are available: Thermophoresis with T jump; Thermophoresis; T jump; and Manual. In the manual mode you can manually define the before/after regions that are to be used in the analysis. Further explanation and discussion of the different modes can be found in Scheuermann et al. (2016). - Inspect the binding curve. The amplitude of the curve should be at least 5 response units and the noise in the baseline should be at least 3 times less than the amplitude.
- To compare several datasets, the fluorescence of each dataset may be normalised, △Fnorm, by selecting this option in the MO.Affinity Analysis program. The normalised fluorescence of each dataset can, in turn, be divided by the amplitude of the binding curve, as seen in Figure 4. We transferred the data points and the associated uncertainties as well as the fitted traces from the MO.Affinity Analysis program to Origin, where the △Fnorm-values were divided by the amplitude of the binding curve and the curves were re-plotted (Figure 4).
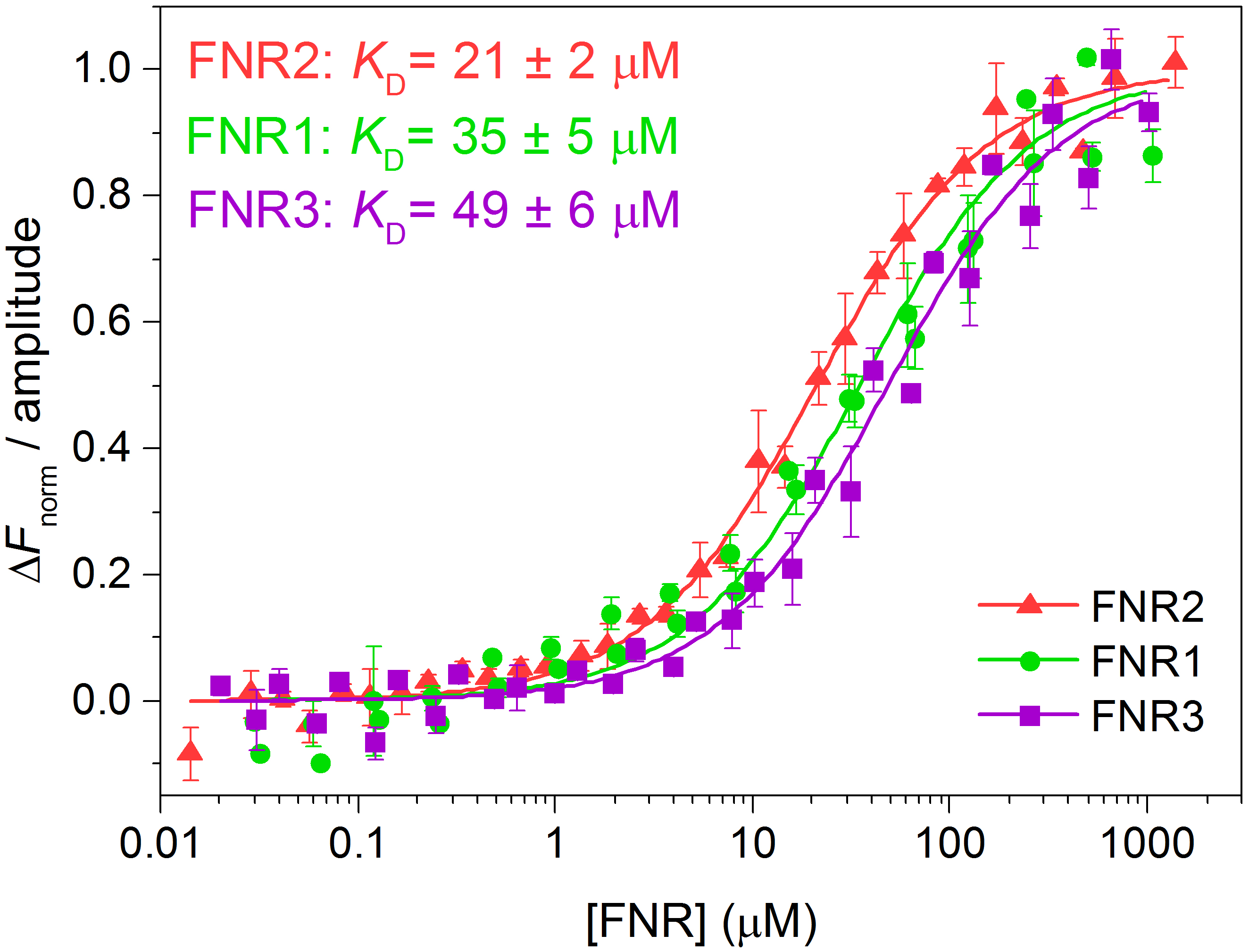
Figure 4. Normalised fluorescence traces (adapted from Lofstad et al., 2016)
- Select the appropriate binding model. We selected KD.
Notes
- The binding constants may vary from one protein preparation to another.
- Valuable control experiments that should be carried out include testing the binding affinities at different pH-values and at different ionic strengths, and also testing the fluorescently labelled protein against either a known non-binding protein or against a protein that is known to bind strongly to the labelled protein (or both).
- Determining the degree of labelling (as described in the FAQ: How do I determine the protein concentration after labeling and the degree of labeling (DOL)? [NanoTemper]):
- Collect an absorbance spectrum of the labelled protein using a UV-vis spectrophotometer.
- Read the absorbance at 280 nm and the absorbance at λmax of the dye (varies depending on the dye used).
- The concentration of the labelled protein can be found using the following formula:
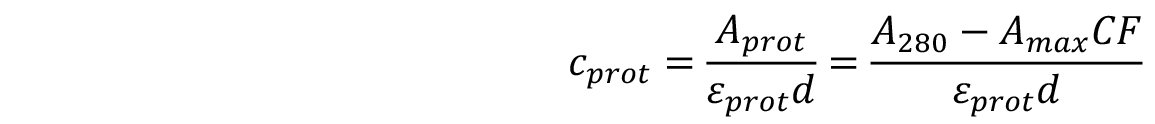
where,
d is the path length of the spectrophotometer,
εprot is the extinction coefficient of the protein,
Amax is the absorbance at λmax,
CF is the correction factor of the dye, taking into account the absorption of the dye at 280 nm.
The degree of labelling can be found using the equation below: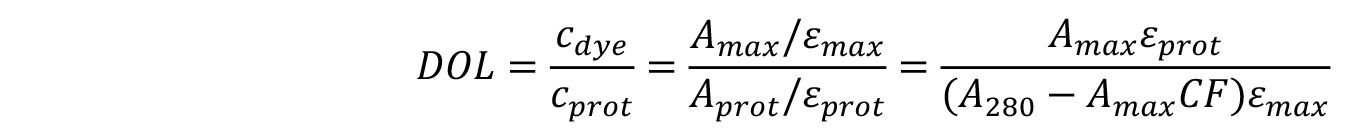
where,
εmax is the extinction coefficient of the dye.
Recipes
- Buffer A (50 mM HEPES, 100 mM KCl, 0.05% Tween-20)
1.192 g HEPES
0.745 g KCl
Make up to 90 ml with Milli-Q distilled H2O, adjust the pH to 7.5 using KOH, and make up to final volume 100 ml with Milli-Q distilled H2O. If necessary, adjust the pH again
Sterile filter the solution using a syringe and 0.22 μm filter and store at 4 °C until use (the buffer has been used within 5 days)
Add 500 μl 10% Tween-20 to a final concentration of 0.05% prior to the experiments
Acknowledgments
This protocol is based on the user manual supplied with the MonolithTM NT.115 instrument (NanoTemper) and the FAQ: How do I determine the protein concentration after labeling and the degree of labeling (DOL)? (NanoTemper). This work is funded by the Norwegian Research Council (Projects 231669 and 214239). The MST instrument is operated with the financial support of the South-Eastern Norway Regional Health Authority (Grant 2015095; Regional Core Facility for Structural Biology). A brief description of this protocol has previously been published in Lofstad et al. (2016).
References
- Duhr, S. and Braun, D. (2006). Why molecules move along a temperature gradient. Proc Natl Acad Sci U S A 103(52): 19678-19682.
- Lofstad, M., Gudim, I., Hammerstad, M., Rohr, A. K. and Hersleth, H.-P. (2016). Activation of the class Ib ribonucleotide reductase by a flavodoxin reductase in Bacillus cereus. Biochemistry 55(36): 4998-5001.
- Røhr, Å. K., Hersleth, H.-P. and Andersson, K. K. (2010). Tracking flavin conformations in protein crystal structures with Raman spectroscopy and QM/MM calculations. Angew Chem Int Ed Engl 49(13): 2324-2327.
- Scheuermann, T. H., Padrick, S. B., Gardner, K. H. and Brautigam, C. A. (2016). On the acquisition and analysis of microscale thermophoresis data. Anal Biochem 496: 79-93.
- Seidel, S. A., Dijkman, P. M., Lea, W. A., van den Bogaart, G., Jerabek-Willemsen, M., Lazic, A., Joseph, J. S., Srinivasan, P., Baaske, P., Simeonov, A., Katritch, I., Melo, F. A., Ladbury, J. E., Schreiber, G., Watts, A., Braun, D. and Duhr, S. (2013). Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 59(3): 301-315.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Gudim, I., Lofstad, M., Hammerstad, M. and Hersleth, H. (2017). Measurement of FNR-NrdI Interaction by Microscale Thermophoresis (MST). Bio-protocol 7(8): e2223. DOI: 10.21769/BioProtoc.2223.
Category
Biochemistry > Protein > Interaction > Protein-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.