- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Differential Salt Fractionation of Nuclei to Analyze Chromatin-associated Proteins from Cultured Mammalian Cells
Published: Vol 7, Iss 6, Mar 20, 2017 DOI: 10.21769/BioProtoc.2175 Views: 17089
Reviewed by: Andrea PuharTatsuki KunohDavid A. Cisneros
Original research article
The authors used this protocol in:

Jul 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Nucleosomes are the core units of cellular chromatin and are comprised of 147 base pairs (bp) of DNA wrapped around an octamer of histone proteins. Proteins such as chromatin remodelers, transcription factors, and DNA repair proteins interact dynamically with chromatin to regulate access to DNA, control gene transcription, and maintain genome integrity. The extent of association with chromatin changes rapidly in response to stresses, such as immune activation, oxidative stress, or viral infection, resulting in downstream effects on chromatin conformation and transcription of target genes. To elucidate changes in the composition of proteins associated with chromatin under different conditions, we adapted existing protocols to isolate nuclei and fractionate cellular chromatin using a gradient of salt concentrations. The presence of specific proteins in different salt fractions can be assessed by Western blotting or mass spectrometry, providing insight into the degree to which they are associated with chromatin.
Keywords: ChromatinBackground
Many chromatin-associated proteins are insoluble under low salt conditions because of their charged-based interaction with DNA or histones. Since salt disrupts charged-based protein-DNA and protein-protein interactions, chromatin-associated proteins become more soluble with increasing concentration of NaCl (Teves and Henikoff, 2012). Proteins strongly bound to DNA are expected to elute with high salt whereas loosely bound proteins, such as transcription factors, will elute with low salt. We are specifically interested in how virus infection alters the composition of factors associated with the cellular chromatin. Nuclear replicating viruses, such as adenovirus, herpes simplex virus, and Epstein-Barr virus, dramatically alter the appearance of the host chromatin during infection (Avgousti et al., 2016; Lam et al., 2010; Simpson-Holley et al., 2005; Chiu et al., 2013). We hypothesized that these changes in appearance are partly due to differences in protein composition of host chromatin. Changes in host chromatin could reflect antiviral defenses mounted by the cell or active manipulation by the virus. To compare association of proteins with chromatin in uninfected and infected cells we developed this protocol to fractionate nuclei using a salt gradient (Figure 1). In this protocol we isolate nuclei, digest the DNA down to mono-nucleosome length, and then wash the nuclei with increasing concentrations of salt, collecting each fraction for analysis by Western blotting. We recently used this protocol to elucidate changes to cellular chromatin during infection with adenovirus (Avgousti et al., 2016). We now present this protocol as a general approach to monitor association of proteins with chromatin under a wide range of perturbing conditions. 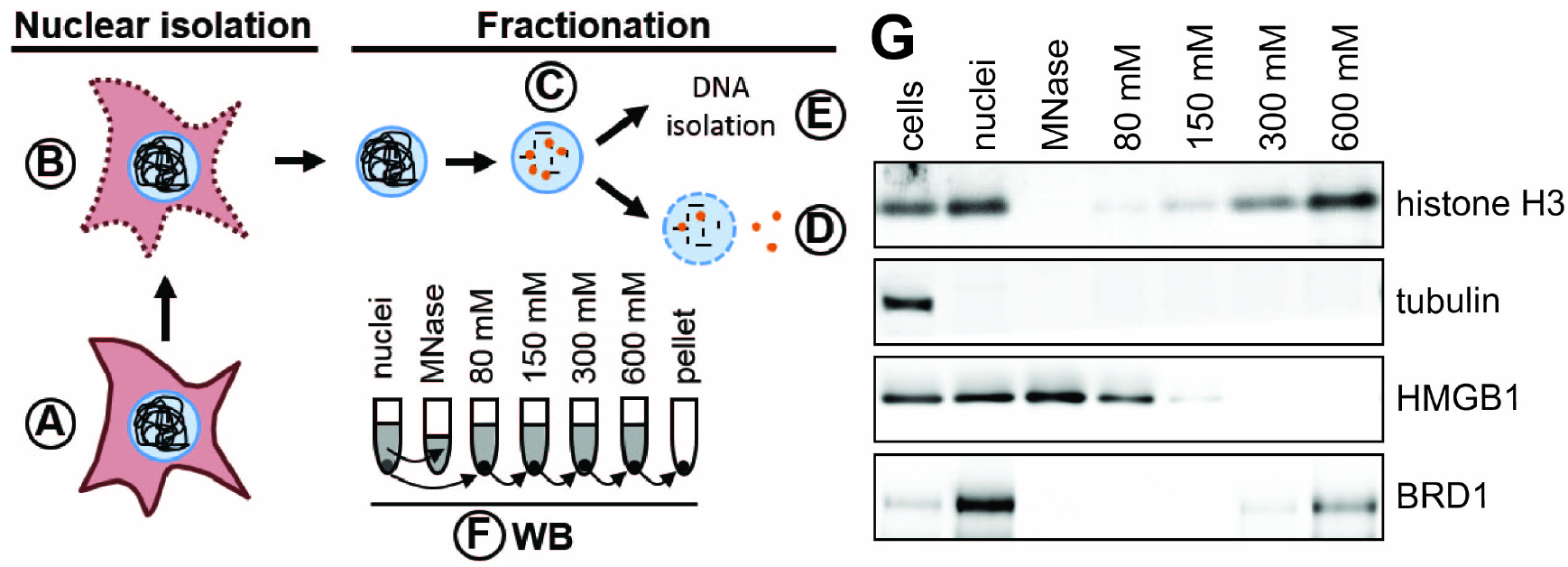
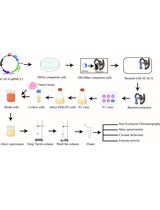
Figure 1. Schematic of nuclear fractionation and example Western blot. A. Roughly 4 x 107 cells are prepared per condition. B. Plasma membranes are permeabilized and nuclei are isolated either by sucrose cushion (step B1) or using a Dounce homogenizer (step B2). C. DNA is digested to mono-nucleosome length using MNase. Proteins loosely bound to chromatin elute during this step. D. The chromatin is further fractionated by washing the nuclei in buffers with increasing salt concentration. E. The DNA is isolated from nuclei to confirm digestion of the cellular genome to 150 bp fragments. F. The quality of fractionation is tested using SDS-PAGE and Western blot (WB) for control proteins (e.g., tubulin, histone H3). The grey colored supernatants (and the pellet in case of the nuclei) represent the samples used for Western blot analysis. G. Example Western blot analysis of chromatin fractionation. Tubulin is found only in the cytoplasmic fraction and is a suitable control to test the quality of nuclear isolation. Histone H3 is a component of cellular chromatin and only elutes from nuclei in buffers with high salt. HMGB1 is a highly mobile nuclear protein (Sapojnikova et al., 2005) and thus elutes during MNase digest and under lower salt conditions. Brd1 directly binds to histone tails (Sanchez et al., 2014) and elutes under high salt conditions.
Materials and Reagents
Note: Comparable reagents from different suppliers may be used for the protocol.
- 150 mm tissue culture dishes (Corning, Falcon®, catalog number: 353025 )
- 15 ml centrifuge tube (Corning, catalog number: 430790 )
- 5 ml pipettes (VWR, catalog number: 89130-908 )
- Transfer pipette (Denville Scientific, catalog number: P7222 )
- 30 ml glass tube (Corning, Corex®, catalog number: 1-8445-30 )
Note: This product has been discontinued. - 1.7 ml microcentrifuge tubes (VWR, catalog number: 87003-294 )
- Pipette tips
0.1-10 µl (Corning, catalog number: 4153 )
1-200 µl (Corning, catalog number: 4126 )
100-1,000 µl (Corning, catalog number: 4129 ) - 250 ml sterile disposable filter units with 0.2 µm PES membrane (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 568-0020 ) (used for Buffer I and Buffer II)
- 60 ml syringe (BD, catalog number: 309653 ) (used for Buffer IV.80, IV.150, IV.300 and IV.600)
- 25 mm syringe filter (Pall, catalog number: 4612 ) (used for Buffer IV.80, IV.150, IV.300 and IV.600)
- A549 cells (ATCC, catalog number: CCL-185 )
- Ham’s F-12K cell culture media (Thermo Fisher Scientific, GibcoTM, catalog number: 21127-022 )
- Fetal bovine serum (FBS) (VWR, catalog number: 89510-182 )
- Penicillin-streptomycin (Pen/Strep) (Thermo Fisher Scientific, GibcoTM, catalog number: 15140-122 )
- Trypsin-EDTA (0.25%) (Thermo Fisher Scientific, GibcoTM, catalog number: 25200-056 )
- Phosphate buffered saline (PBS) (Mediatech, catalog number: 21-030-CM )
- Liquid nitrogen
- NP-40/IGEPAL® CA-630 (Sigma-Aldrich, catalog number: I8896 ) (10% stock solution in H2O)
- Phenylmethanesulfonyl fluoride (PMSF) (Sigma-Aldrich, catalog number: P7626 ) (0.1 M stock solution in isopropanol)
- 1,4-dithiothreitol (DTT) (Sigma-Aldrich, catalog number: 10197777001 ) (1 M stock solution in HEPES buffer, pH 7.75)
- Protease inhibitor cocktail (Roche Diagnostics, catalog number: 11697498001 ) (prepared as 50x stock solution in H2O according to manufacturer instructions)
- Micrococcal nuclease (MNase) (Sigma-Aldrich, catalog number: N3755 ) (0.2 U/µl stock solution in H2O)
- Ethylene glycol-bis(2-aminoethylether)-N,N,N’,N’-tetraacetic acid (EGTA) (Sigma-Aldrich, catalog number: E3889 ) (0.1 mM stock solution in H2O, pH = 10)
- PCR purification kit (QIAGEN, catalog number: 28104 )
- Orange G (Sigma-Aldrich, catalog number: O3756 ) (0.35% [w/v] orange G with 30% [w/v] sucrose in H2O for 6x stock solution)
- 100 bp DNA ladder (New England Biolabs, catalog number: N3231 )
- Broad range protein ladder (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 26623 )
- GelRed nucleic acid gel stain (Biotum, catalog number: 41003 )
- LDS sample buffer (4x) (Thermo Fisher Scientific, NovexTM, catalog number: NP0007 )
- Sucrose (Fisher Scientific, catalog number: BP220-1 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P9541 ) (1 M stock solution in H2O)
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S9625 ) (5 M stock solution in H2O)
- Magnesium chloride hexahydrate (MgCl2·6H2O) (Sigma-Aldrich, catalog number: M2670 ) (1 M stock solution in H2O)
- Trizma base (Sigma-Aldrich, catalog number: T1503 ) (1 M stock solution in H2O adjusted to pH 7.4 with HCl)
- UltraPure agarose (Thermo Fisher Scientific, InvitrogenTM, catalog number: 16500500 )
- Hydrochloric acid 6.0 N solution (HCl) (Fisher Scientific, catalog number: MK-H168-4 )
- Calcium chloride dihydrate (CaCl2·2H2O) (Sigma-Aldrich, catalog number: C5080 ) (0.5 M stock solution in H2O)
- Triton X-100 (Sigma-Aldrich, catalog number: T8787 )
- HEPES (Sigma-Aldrich, catalog number: H3375 ) (1 M stock solution in H2O adjusted to pH 7.9 with NaOH)
- Sodium hydroxide (NaOH) (AMRESCO, catalog number: M137 )
- Buffer I.A and I.B (see Recipes)
- Buffer II (see Recipes)
- Buffer III.A and III.B (see Recipes)
- Buffer IV.80, IV.150, IV.300 and IV.600 (see Recipes)
- Hypotonic buffer (see Recipes)
Equipment
Note: Equipment with similar properties may be used for the protocol, however, we recommend using a specific kind of reusable centrifuge tubes (listed in 5) to ensure high quality isolation of nuclei.
- CO2 incubator for cell culture (BINDER, catalog number: 9040-0082 )
- Benchtop centrifuge (Beckman Coulter, model: Allegra X-14R )
- Rotors for benchtop centrifuge (Beckman Coulter, models: SX4750 for tissue culture and FX6100 for 10,000 x g spins, or seminal rotors suitable for high speeds)
- Adapters for FX6100 rotor (Beckman Coulter, catalog number: 392830 )
- 30 ml reusable centrifuge tubes (Sigma-Aldrich, catalog number: T2793 )
- Tabletop centrifuge 5424 R (Eppendorf, model: 5424 R )
- 1 ml tissue grinder (Dounce homogenizer) with tight fitting pestle (Ace Glass Incorporated, catalog number: 8343-01 )
- Water bath (Fisher Scientific, model: IsotempTM Digital-Control Water Baths Model 215 , catalog number: 15-462-15Q)
- Tube rotator (VWR, catalog number: 10136-084 )
- Pipettes
1-10 µl (Gilson, catalog number: F144055P )
2-20 µl (Gilson, catalog number: F144056M )
20-200 µl (Gilson, catalog number: F144058M )
100-1,000 µl (Gilson, catalog number: F144059M ) - Agarose gel electrophoresis systems (Thermo Fisher Scientific, Thermo ScientificTM, model: Owl EasyCast B1A system )
- Fluorescence and chemiluminescence gel imaging system (Syngene, model: G: BOX Chemi XT4 )
- Heat block (Fisher Scientific, model: IsotempTM Digital Dry Baths/Block Heaters , catalog number: 88-860-022)
- Protein electrophoresis apparatus (Bio-Rad Laboratories, model: Mini-PROTEAN® Tetra Vertical Electrophoresis Cell for Mini Precast Gels , catalog number: 1658005)
- Western blot apparatus (Thermo Fisher Scientific, model: SureLockTM Mini-Cell Electrophoresis System )
Software
- ImageJ (freely available from National Institutes of Health, https://imagej.nih.gov/ij/)
Procedure
- Cell culture
Note: This protocol was optimized for nuclear isolation and chromatin fractionation of roughly 4 x 107 A549 cells (for A549 cells that is approximately two 100% confluent 150 mm cell culture dishes). This number of cells was chosen for efficient nuclear isolation as described in Procedure B. This protocol can be used for other cell types but should be optimized accordingly.- For each condition, grow roughly 4 x 107 A549 cells in F-12K media with 10% FBS and 1% Pen/Strep.
Note: Time treatments such that the required cell number is reached at the time of harvest. - Harvest cells using trypsin and combine into one 15 ml tube per condition.
- Centrifuge for 2 min, 500 x g, room temperature using benchtop centrifuge (Allegra X-14R with rotor SX4750).
- Wash cells once with 10 ml of PBS and centrifuge again for 2 min, 500 x g, room temperature.
- Aspirate as much of the supernatant as possible without disturbing the cell pellet.
- Flash freeze samples in liquid nitrogen.
- Store samples at -80 °C until ready to process.
- For each condition, grow roughly 4 x 107 A549 cells in F-12K media with 10% FBS and 1% Pen/Strep.
- Nuclear isolation
Note: This section describes two alternative methods for nuclear isolation. The procedure detailed in B1 yields a clean nuclear fraction but B2 is recommended for highly mobile nuclear proteins that are lost from the nuclear fraction when mild detergents are used as described in B1 (see example in Figure 2).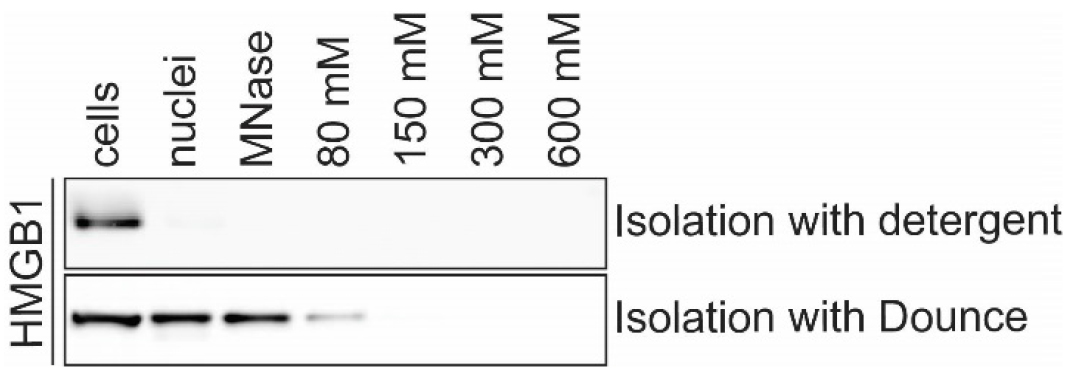
Figure 2. Fractionation of nuclear protein HMGB1 can differ depending on the method of nuclear isolation. Western blot of chromatin fractionation of A549 cells using different methods of nuclear isolation. HMGB1 is lost from nuclei during nuclear isolation B1 using NP-40 and a sucrose cushion. HMGB1 is retained in the nuclear fraction during isolation of nuclei with the alternative method described in step B2.- Nuclear isolation using mild detergent and sucrose cushion.
Note: This section describes permeabilizing the cell membrane using a mild detergent (NP-40), followed by separation of the nuclei from cytoplasmic debris by sucrose cushion. The sucrose cushion consists of a layer with 1.2 M sucrose at the bottom (Buffer II) and a layer with 0.32 M sucrose on top (Buffer I, Figure 3A). The nuclei are more dense than the 1.2 M sucrose buffer and will pellet at the bottom of the tube while the less dense cytoplasmic debris will remain in the upper sucrose layer (Figure 3B).- Thaw cells on ice (about 10 min until pellet is loose).
- In the meantime, prepare one aliquot each of Buffer I.A and I.B by addition of PMSF, DTT, and protease inhibitor cocktail immediately before use (see Recipes for final concentrations). Add NP-40 to Buffer I.B.
- Gently resuspend cells in 2 ml of Buffer I.A using a 5 ml pipette. Avoid the formation of bubbles.
- Set aside 50 μl of the cell suspension in a new tube labeled ‘cells fraction’.
- Add 2 ml of Buffer I.B containing NP-40 to the 2 ml of cell suspension and mix gently by inverting the tube 2-3 times.
- Incubate samples on ice for 10 min. Mix gently by inverting after 5 min.
- Pipette 8 ml of ice-cold Buffer II into 30 ml reusable centrifuge tubes (one per sample).
- Carefully layer cell suspension onto Buffer II (Figure 3A).
- If the number of tubes is even, balance the tubes with Buffer I.A before centrifugation. If the number of tubes is uneven, use an additional tube containing water as balance.
- Centrifuge for 20 min, 10,000 x g, 4 °C using benchtop centrifuge (Allegra X-14R with rotor FX6100) with low brakes (deceleration setting 1 for this centrifuge model, 5-10 min of deceleration for other centrifuge models) to avoid disruption of pellet.
Note: Use the adapters listed in the Equipment section under 4 to fit the 30 ml reusable centrifuge tubes into the FX6100 rotor. The nuclei form a dense pellet at the bottom of the tube while the cytoplasmic debris remains in the upper sucrose layer, Figure 3C. - Carefully remove the supernatant using a transfer pipette. Keep pellet containing the nuclei and proceed to Procedure C.

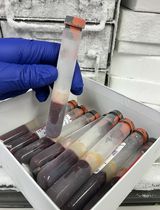
Figure 3. Intermediate steps of nuclear isolation. A. Layers of the sucrose cushion. 1 = cells resuspended in Buffer I containing 0.32 M sucrose. 2 = Buffer II containing 1.2 M sucrose. Clear glass tube was used for better visibility of the two layers. B. Layers of sucrose cushion as seen before the centrifugation step in the 30 ml reusable tube. Layer 1 = Buffer I. Layer 2 = Buffer II. C. Layers of sucrose cushion as seen after the centrifugation step in the 30 ml reusable tube. Layer 1 = Buffer I. Layer 2 = Buffer II. D and E. Pellet of nuclei after spin at 10,000 x g indicated by red arrow. The buffers were removed before these pictures were taken.
- Thaw cells on ice (about 10 min until pellet is loose).
- Nuclear isolation by manual disruption
Note: Using mild detergents such as NP-40 for nuclear isolation is important to ensure that the nuclear membrane remains largely intact. In some cases, highly mobile nuclear proteins such as HMGB1 (Sapojnikova et al., 2005) may diffuse out of the nucleus upon isolation with this method. To prevent loss of these proteins from the nuclear fraction, we adapted an alternative method that relies on a hypotonic buffer and subsequent manual disruption of the cell membrane using a Dounce homogenizer. The short centrifugation times in this step minimize diffusion of these proteins out of the nuclei (Figure 2).- Thaw cells on ice (about 10 min until pellet is loose).
- Gently resuspend cells in 1 ml of hypotonic buffer and transfer into a 1.7 ml tube using a P1000 pipette with the tip cut (or wide orifice pipette tips) to avoid disrupting the cellular membranes.
- Set aside 25 μl of the cell suspension in a new tube as ‘cells fraction’.
Note: This is proportional to the 50 μl aliquot taken from 2 ml of cell suspension in step B1. - Incubate on ice for 30 min.
- Pre-cool the Dounce homogenizer and the pestle on ice for at least 5 min.
- Transfer the cell suspension to cold Dounce homogenizer using a transfer pipette.
- Disrupt the cell membrane using 40 strokes of the tight-fitting pestle. Minimize the formation of bubbles.
- Transfer sample to 1.7 ml tube using a transfer pipette.
- Centrifuge for 5 min, 1,500 x g, 4 °C using a tabletop centrifuge.
- Carefully remove the supernatant using a P1000 pipette. Transfer supernatant and label as ‘cytosol fraction’ if desired. Keep pellet containing the nuclei and proceed to Procedure C.
Note: The nuclei form a loose pellet at the bottom of the tube that can be easily disturbed. The cytoplasmic fraction in step B1 is too dilute to isolate as a result of the sucrose cushion.
- Thaw cells on ice (about 10 min until pellet is loose).
- Nuclear isolation using mild detergent and sucrose cushion.
- Micrococcal nuclease digestion
Note: In this step the DNA is digested to the length of mono-nucleosomes (roughly 150 bp) using MNase. While partial MNase digest in combination with nuclear fractionation by salt gradient can be used to separate euchromatin from heterochromatin (Teves and Henikoff, 2012), our protocol aims to identify global association of proteins with chromatin and does not distinguish between different types of chromatin. Subsequently, it is important that the DNA is completely digested into mono-nucleosomes to break up chromatin and allow for elution of all soluble proteins in Procedure D (Figure 4). EGTA is used instead of EDTA to stop the MNase digestion as EGTA preferentially chelates Ca2+ ions that are necessary for MNase enzymatic activity but does not impact Mg2+ ions important for protein-protein interactions. Proteins loosely bound to chromatin elute during MNase digest and can be detected in the supernatant collected in this section of the protocol.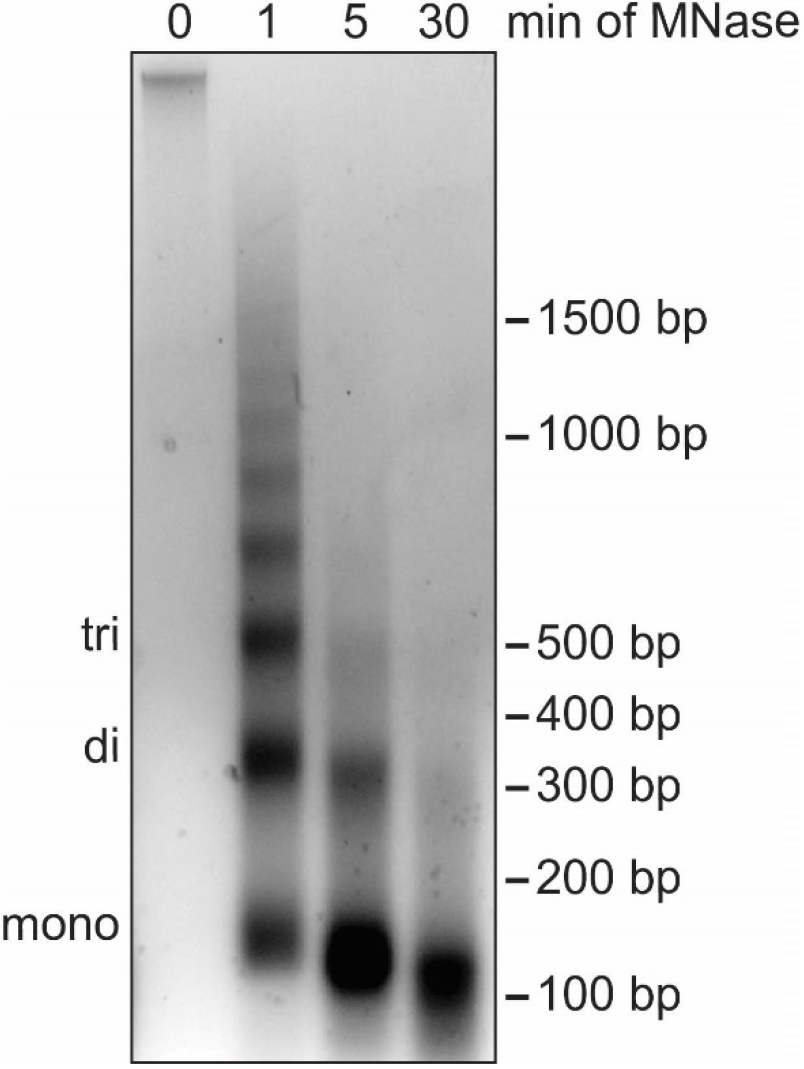
Figure 4. Digestion of cellular DNA using MNase. Agarose gel analysis of DNA extracted from nuclei during a time course of MNase treatment. The position of DNA bands corresponding to mono-, di- and tri-nucleosomes are indicated on the left while the DNA size in base pairs (bp) is indicated on the right. Without MNase digestion (0 min), cellular DNA barely enters the agarose gel because of the large size of the DNA molecules. After 1 min of MNase digestion, DNA fragments show the characteristic banding pattern of multiples of 150 bp. After 5 min the DNA fragments correspond to one to three nucleosomes in length. After 30 min most DNA fragments are close to 150 bp, indicating the desired digestion of cellular DNA necessary for optimal chromatin fractionation. The high concentration of DNA causes the slightly lower shift of the mono-nucleosome band. The nuclei used for this time course were isolated using method B.1. No differences in MNase digestion efficiency have been observed with nuclei isolated using method B.2.- Prepare and cool Buffer III.A and III.B.
- Add 400 μl Buffer III.A to the nuclei.
- Gently resuspend the nuclei using a P1000 pipette with the tip cut (or wide orifice pipette tips) to avoid disrupting the nuclear membranes.
- Add 5 μl of MNase (1 U) and incubate at 37 °C (in a water bath) for 30 min.
- Mix every 10 min by gently inverting the tube 2-3 times.
- After 30 min, add 25 μl of ice-cold 0.1 M EGTA to stop the digest.
- Set aside a 60 μl aliquot of the nuclear suspension as ‘nuclei fraction’ and for DNA isolation to confirm the sufficient digest of DNA (described in Procedure E).
- Centrifuge for 10 min, 400 x g, 4 °C.
- Transfer supernatant to fresh tube and label ‘MNase fraction’.
- Wash the nuclei once by resuspending them in 400 μl Buffer III.B.
- Centrifuge again for 10 min, 400 x g, 4 °C. Discard the supernatant.
- Prepare and cool Buffer III.A and III.B.
- Chromatin fractionation
Note: In this step the nuclei are further fractionated using buffers with increasing amounts of NaCl. Proteins only weakly bound to DNA are expected to be soluble under low salt conditions and will elute from nuclei in the buffers with low NaCl concentration. Proteins tightly bound to chromatin only become soluble under high salt conditions and only elute from nuclei in the buffers with high NaCl concentration.- Gently resuspend the nuclei in 400 μl Buffer IV.80 using a P1000 pipette with the tip cut (or wide orifice pipette tips).
- Rotate at 4 °C for 30 min.
- Centrifuge for 10 min, 400 x g, 4 °C.
- Transfer supernatant to fresh tube and label ‘80 mM fraction’.
- Repeat steps D1-D4 with Buffer IV.150, IV.300 and IV.600 in that order. Keep the supernatants as fractions ‘150 mM’, ‘300 mM’ and ‘600 mM’, respectively.
- Prepare samples for Western blot immediately (see Table 1 in Procedure F) or store at -20 °C.
- Gently resuspend the nuclei in 400 μl Buffer IV.80 using a P1000 pipette with the tip cut (or wide orifice pipette tips).
- DNA isolation and DNA gel
Note: In this step, DNA is isolated from the samples after MNase digestion to test the length of DNA fragments. The goal is to digest the DNA down to mono-nucleosome level of around 150 bp to ensure optimal chromatin fractionation (Figure 4). It is important that orange G or another small molecular weight dye is used for loading the DNA. Commonly used DNA loading dyes such as bromophenol blue run around the same size as 150 bp of DNA and may obscure the results.- Dilute 10 μl of the ‘nuclei’ fraction in 90 μl H2O for better DNA purification.
- Use a PCR purification kit to isolate the DNA from this sample following the kit instructions. Elute the DNA in 30 μl of H2O.
- Take out 5 μl (about 5 μg) of DNA and add 1 μl of 6x Orange G. Keep the rest of the DNA on ice until results are finalized or freeze at -20 °C.
- Run samples and a 100 bp ladder on a 2% agarose gel containing 1x GelRed at 100 V for 1 h.
- Visualize the DNA using gel imager and check for prominent DNA band of around 150 bp with minimal DNA laddering (Figure 4).
- Dilute 10 μl of the ‘nuclei’ fraction in 90 μl H2O for better DNA purification.
- Verifying the quality of nuclear fractionation by Western blot
Note: The quality of the nuclear fractionation can be analyzed using SDS-PAGE combined with Western blotting (Figure 1G). Efficient nuclear isolation can be tested by probing for proteins such as tubulin that are only found in the cytoplasm. The quality of chromatin fractionation can be tested by probing for histone proteins such as histone H3 or proteins expected to bind histone tails such as bromodomain containing protein Brd1 (Sanchez et al., 2014). The interaction of the histone H3 and Brd1 with chromatin should only be disrupted under high salt conditions. In addition, proteomics can be performed to identify changes to global chromatin composition under different conditions. Further details concerning mass spectrometry of salt fractions can be found in (Avgousti et al., 2016).- Prepare samples for analysis by Western blotting according to Table 1.
Table 1. Preparation of different fractions for Western blotFraction Sample H2O 4x LDS sample buffer with 10% DTT cells (from B1) 10 μl 20 μl 10 μl cells (from B2) 5 μl 25 μl 10 μl nuclei 10 μl 20 μl 10 μl MNase 30 μl - 10 μl 80 mM 30 μl - 10 μl 150 mM 30 μl - 10 μl 300 mM 30 μl - 10 μl 600 mM 30 μl - 10 μl - Denature samples at 95 °C (in a heat block) for 10 min.
- Analyze 15 μl of each sample via SDS-PAGE and Western blot. Probe for tubulin and histone H3 as controls.
- Prepare samples for analysis by Western blotting according to Table 1.
Data analysis
To compare the association of a protein of interest with chromatin under different conditions, the samples should be run on the same SDS-PAGE gel and Western blot as different exposures may confound the interpretation. Expected results for various proteins are described here to aid in data analysis, though each protein of interest tested should be considered separately. The ‘cells’ fraction represents the total amount of protein in a sample for relative comparison with other fractions. A cytoplasmic protein such as tubulin should only be present in the ‘cells’ fraction (Figure 1G). Nuclear proteins should also be present in the ‘nuclei’ fraction as seen for H3 and Brd1 (Figure 1G). The band intensity for the remaining fractions represents the solubility of nuclear proteins under these conditions. H3 and Brd1 have the highest band intensity in the ‘600 mM’ fraction, signifying greater solubility under high salt conditions typical for chromatin-associated proteins. For HMGB1 the highest band intensity can be observed for the ‘MNase’ and ‘80 mM’ fraction, indicating solubility under low salt conditions, suggesting weak association with chromatin. Differences in band intensity for different fractions can be quantified using ImageJ (freely available from National Institutes of Health, https://imagej.nih.gov/ij/).
Details concerning data analysis for mass spectrometry of salt fractions can be found in (Avgousti et al., 2016).
Notes
This protocol is suitable to compare changes in the chromatin-associated proteome under different conditions. We have used this method to show changes to cellular chromatin during infection with adenovirus (Avgousti et al., 2016).
Recipes
- Buffer I.A and I.B
0.32 M sucrose
60 mM KCl
15 mM NaCl
5 mM MgCl2
0.1 mM EGTA
15 mM Tris pH 7.4
Filtered and stored at 4 °C
Add fresh:
0.5 mM DTT
0.1 mM PMSF
1x protease inhibitor cocktail
Add only to Buffer I.B:
0.1% NP-40 - Buffer II
1.2 M sucrose
60 mM KCl
15 mM NaCl
5 mM MgCl2
0.1 mM EGTA
15 mM Tris pH 7.4
Filtered and stored at 4 °C
Add fresh:
0.5 mM DTT
0.1 mM PMSF
1x protease inhibitor cocktail - Buffer III.A and III.B
10 mM Tris pH 7.4
2 mM MgCl2
0.1 mM PMSF
Add only to Buffer III.A:
5 mM CaCl2 - Buffer IV.80, IV.150, IV.300 and IV.600
10 mM Tris pH 7.4
2 mM MgCl2
2 mM EGTA
0.1% Triton X-100
Add the following concentrations of NaCl to the individual buffers:
70 mM Buffer IV.80
140 mM Buffer IV.150
290 mM Buffer IV.300
590 mM Buffer IV.600
Filtered and stored at 4 °C
Add fresh:
0.1 mM PMSF - Hypotonic buffer
10 mM HEPES pH 7.9
1.5 mM MgCl2
10 mM KCl
0.1 mM PMSF
0.5 mM DTT
Acknowledgments
We thank members of the Weitzman laboratory for carefully reading and revising the protocol. This work was supported by a grant from the National Institutes of Health (CA097093), the Institute for Immunology of the University of Pennsylvania, and funds from the Children’s Hospital of Philadelphia (M.D.W.). D.C.A. was supported in part by T32 CA115299 and F32 GM112414. The protocol described herein was published in Avgousti et al., 2016.
References
- Avgousti, D. C., Herrmann, C., Kulej, K., Pancholi, N. J., Sekulic, N., Petrescu, J., Molden, R. C., Blumenthal, D., Paris, A. J., Reyes, E. D., Ostapchuk, P., Hearing, P., Seeholzer, S. H., Worthen, G. S., Black, B. E., Garcia, B. A. and Weitzman, M. D. (2016). A core viral protein binds host nucleosomes to sequester immune danger signals. Nature 535(7610): 173-177.
- Chiu, Y. F., Sugden, A. U. and Sugden, B. (2013). Epstein-Barr viral productive amplification reprograms nuclear architecture, DNA replication, and histone deposition. Cell Host Microbe 14(6): 607-618.
- Lam, Y. W., Evans, V. C., Heesom, K. J., Lamond, A. I. and Matthews, D. A. (2010). Proteomics analysis of the nucleolus in adenovirus-infected cells. Mol Cell Proteomics 9(1): 117-130.
- Sanchez, R., Meslamani, J. and Zhou, M. M. (2014). The bromodomain: from epigenome reader to druggable target. Biochim Biophys Acta 1839(8): 676-685.
- Sapojnikova, N., Maman, J., Myers, F. A., Thorne, A. W., Vorobyev, V. I. and Crane-Robinson, C. (2005). Biochemical observation of the rapid mobility of nuclear HMGB1. Biochim Biophys Acta 1729(1): 57-63.
- Simpson-Holley, M., Colgrove, R. C., Nalepa, G., Harper, J. W. and Knipe, D. M. (2005). Identification and functional evaluation of cellular and viral factors involved in the alteration of nuclear architecture during herpes simplex virus 1 infection. J Virol 79(20): 12840-12851.
- Teves, S. S. and Henikoff, S. (2012). Salt fractionation of nucleosomes for genome-wide profiling. Methods Mol Biol 833: 421-432.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Herrmann, C., Avgousti, D. C. and Weitzman, M. D. (2017). Differential Salt Fractionation of Nuclei to Analyze Chromatin-associated Proteins from Cultured Mammalian Cells. Bio-protocol 7(6): e2175. DOI: 10.21769/BioProtoc.2175.
Category
Immunology > Host defense > General
Cancer Biology > Cancer biochemistry > Protein
Cell Biology > Organelle isolation > Nuclei
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.