- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Multiplexed GuideRNA-expression to Efficiently Mutagenize Multiple Loci in Arabidopsis by CRISPR-Cas9
Published: Vol 7, Iss 5, Mar 5, 2017 DOI: 10.21769/BioProtoc.2166 Views: 12994
Reviewed by: Rainer MelzerMarta BjornsonMoritz BomerDaniel Savatin
Original research article
The authors used this protocol in:

Apr 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Since the discovery of the CRISPR (clustered regularly interspaced short palindromic repeats)-associated protein (Cas) as an efficient tool for genome editing in plants (Li et al., 2013; Shan et al., 2013; Nekrasov et al., 2013), a large variety of applications, such as gene knock-out, knock-in or transcriptional regulation, has been published. So far, the generation of multiple mutants in plants involved tedious crossing or mutagenesis followed by time-consuming screening of huge populations and the use of the Cas9-system appeared a promising method to overcome these issues. We designed a binary vector that combines both the coding sequence of the codon optimized Streptococcus pyogenes Cas9 nuclease under the control of the Arabidopsis thaliana UBIQUITIN10 (UBQ10)-promoter and guide RNA (gRNA) expression cassettes driven by the A. thaliana U6-promoter for efficient multiplex editing in Arabidopsis (Yan et al., 2016). Here, we describe a step-by-step protocol to cost-efficiently generate the binary vector containing multiple gRNAs and the Cas9 nuclease based on classic cloning procedure.
Keywords: CRISPR-Cas9Background
The RNA-guided Cas9-system is derived from the bacterial defense system against foreign DNA (Sorek et al., 2013). It has been recognized as a method of choice for genome editing because of its high efficiency, easy handling and possibility of multiplex editing. In general, the Cas9-gene editing system involves a single synthetic RNA molecule, the gRNA that directs the Cas9 protein to target the desired DNA site for genome modification or transcriptional control. The gRNA-Cas9 complex recognizes the targeted DNA by gRNA-DNA pairing and requires the presence of a protospacer-adjacent motif (PAM). The PAM is represented by the nucleotides NGG or less specific NAG (with N for any nucleotide) in the target site following the gRNA-DNA pairing region. Thus, the approximate 20 nucleotides long gRNA spacer sequence, i.e., the part of the gRNA sequence complementary to the DNA target site, determines the specificity of the complex. In this protocol, we describe the details to generate a binary vector that contains both the gRNA and the Cas9 coding sequence by classic cloning (Figure 1). As our vector system allows for subsequent addition of further gRNAs, it can be used for multiplex editing of the Arabidopsis genome and to obtain multiple, stably inherited alleles. Strong expression of the Cas9 protein and the gRNAs especially in proliferating tissues is achieved by the use of the A. thaliana UBQ10- and the U6-promoter, respectively. First mutations can be detected in the T1 generation, and T-DNA- and Cas9-free mutant plants may already be selected in the T2 generation. Besides the selection of the gRNA and the construction of the plasmid, we give an overview of efficient genotyping methods required for detection of small or large deletions.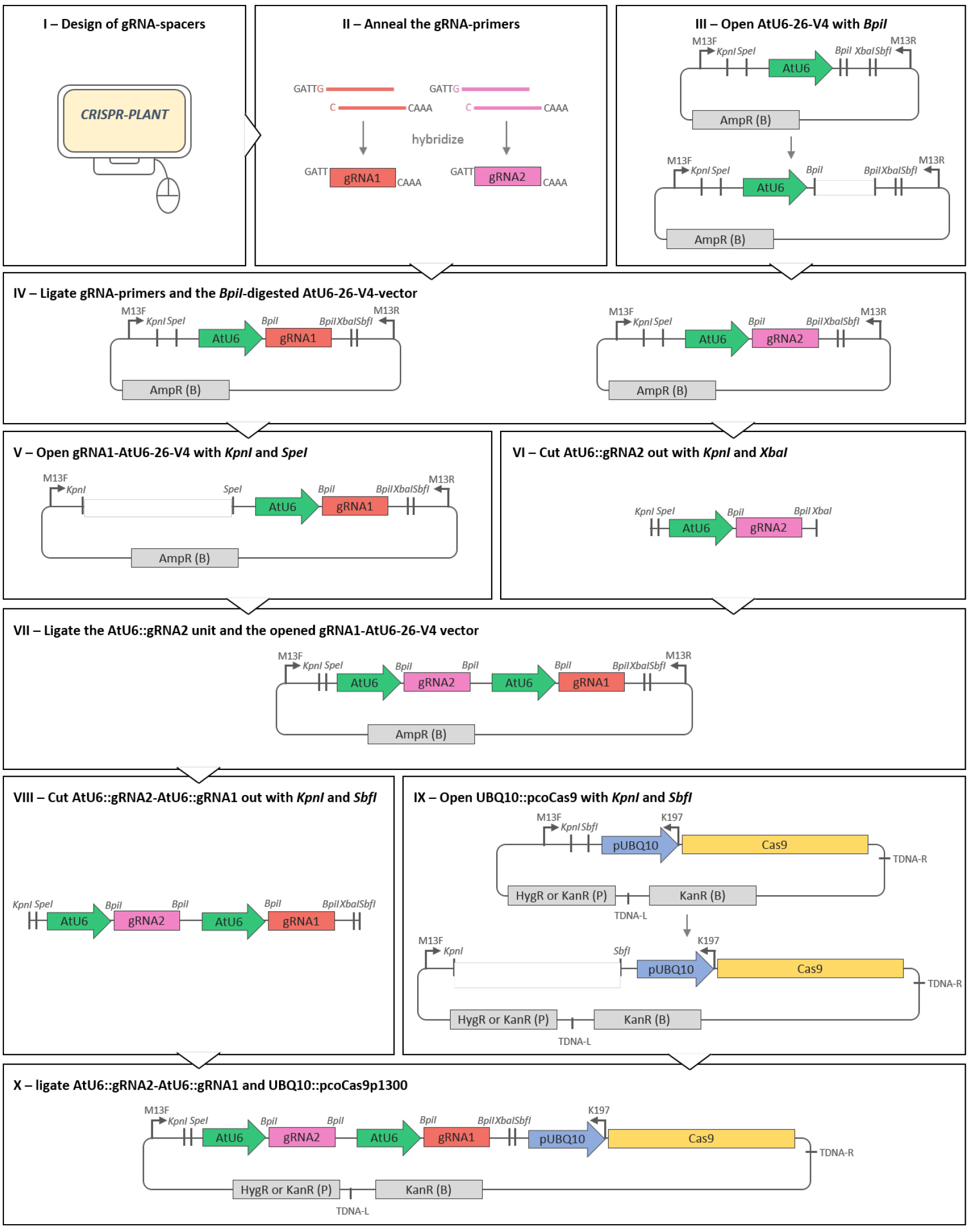
Figure 1. Scheme of the cloning procedure described in the protocol. Only a series of restriction and ligation steps is required to obtain a plant transformation vector equipped with a set of AtU6-driven gRNAs and the Cas9 enzyme under the control of the UBQ10 promoter. Restriction enzyme cutting sites are displayed in italics. Arrows on vectors indicate primer binding sites. TDNA-R and TDNA-L point out T-DNA right and left borders, respectively. (B) and (P) indicate selection markers for selection in bacteria or plants, respectively.
Materials and Reagents
Note: The protocol described here is based on the RNA-guided Cas9 system which was published recently (Yan et al., 2016). Only classical cloning methods such as restriction enzyme digestion and cohesive end ligation are required to construct plasmids ready for plant transformation.
- Consumables
- 200 µl PCR tubes (Kisker Biotech, catalog number: G003-SF )
or 96-well PCR plates (SARSTEDT, catalog number: 72.1978.202 )
StarSeal sealing tape (STARLAB, catalog number: E2796-9793 ) - 1.5 ml reaction tubes (SARSTEDT, catalog number: 72.690.001 )
- Pipette tips
- Scalpel
- Petri dishes, round, 9.2 x 1.6 cm (SARSTEDT, catalog number: 82.1472.001 )
- Petri dishes, square, 10 x 10 x 2 cm (SARSTEDT, catalog number: 82.9923.422 )
- Cuvettes for electroporation, e.g., Gene Pulser® cuvette 0.1 cm (Bio-Rad Laboratories, catalog number: 1652089 )
- 5 ml glass pipette
- MicroporeTM tape (VWR, catalog number: 115-8172 )
Note: Any appropriate consumable can be used. - Competent cells
- One Shot® TOP10 chemically competent Escherichia coli (Thermo Fisher Scientific, InvitrogenTM, catalog number: C4040 )
- Electro-competent Agrobacterium tumefaciens, strain pGV3101 (for preparation of electro-competent Agrobacteria, please see Mersereau et al., 1990)
- Plant material
- Arabidopsis thaliana Col-0
- Plasmids
- AtU6-26-V4 (3.5 kb, ampicillin resistance marker [AmpR], available on request)
- UBQ10::pcoCas9p1300 (14.3 kb, kanamycin resistance marker [KanR] in bacteria, hygromycin or kanamycin resistance marker [HygR or KanR] in plants, available on request)
- Optional: pGEM®-T easy (Promega, catalog number: A3600 )
- Enzymes and buffers
Restriction- BpiI (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: ER1011 )
- KpnI-HF (New England Biolabs, catalog number: R3142 )
- XbaI (New England Biolabs, catalog number: R0145 )
- SpeI-HF (New England Biolabs, catalog number: R3133 )
- SbfI-HF (New England Biolabs, catalog number: R3642 )
- Cutsmart® buffer (New England Biolabs, catalog number: B7204S , supplied with the enzyme)
Genotyping (bacteria) - BpiI (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: ER1011 )
- Green Taq DNA polymerase (GenScript, catalog number: E00043 )
- 10x Taq buffer (GenScript, catalog number: B0005 , supplied with the enzyme)
- 10 mM dNTPs (Carl Roth, catalog number: K039 )
- Any DNA-loading dye, e.g., 6x gel loading dye, purple (New England Biolabs, catalog number: B7024 )
- Any DNA-size standard, e.g., GeneRulerTM 1 kb Plus DNA ladder (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: SM1331 )
- Agarose (Carl Roth, catalog number: 3810 )
- 10x TBE electrophoresis buffer (see Recipes)
Tris (Applichem, catalog number: A1379 )
Boric acid (Applichem, catalog number: 131015 )
EDTA (Applichem, catalog number: 131669 ) - Deionized water (sterile)
- Antibiotics (store stocks at -20 °C)
- Ampicillin (Carl Roth, catalog number: K029 , stock 50 mg/ml in deionized water)
- Kanamycin (Carl Roth, catalog number: T832 , stock 30 mg/ml in deionized water)
- Rifampicin (Applichem, catalog number: A2220 , stock 25 mg/ml in DMSO)
- Gentamycin (Carl Roth, catalog number: 0233 , stock 10 mg/ml in deionized water)
- Hygromycin (Carl Roth, catalog number: CP13 , stock 10 mg/ml in deionized water)
- Media (see Recipes)
- YEB medium for Agrobacterium
Meat extract (Carl Roth, catalog number: 5770 )
Yeast extract (Carl Roth, catalog number: 2904 )
Peptone (Sigma-Aldrich, catalog number: 82303 )
Sucrose (Carl Roth, catalog number: 4621 )
Magnesium sulfate (MgSO4) (Carl Roth, catalog number: P027 )
Bacto-agar (Th. Geyer, CHEMSOLUTE®, catalog number: 9914-500G )
Sodium hydroxide (NaOH) (Merck, catalog number: 28245 ) - YT medium for E. coli
Sodium chloride (NaCl) (Carl Roth, catalog number: 9265 )
Yeast extract (Carl Roth, catalog number: 2904)
Peptone (Sigma-Aldrich, catalog number: 82303)
Bacto-agar (Th. Geyer, CHEMSOLUTE®, catalog number: 9914-500G)
Sodium hydroxide (NaOH) (Merck, catalog number: 28245) - ½ MS for Arabidopsis
MS + B5 Vitamins (Duchefa Biochemie, catalog number: M0231 )
Potassium hydroxide (KOH) (Merck Millipore, catalog number: 105033 ) - DNA purification
- Gel-purification, e.g., NucleoSpin® Gel and PCR clean-up (MACHEREY-NAGEL, catalog number: 740609 )
- Plasmid isolation, e.g., NucleoSpin® Plasmid EasyPure (MACHEREY-NAGEL, catalog number: 740727 )
- Silwet L-77 (Lehle seeds, catalog number: VIS-30 ) for plant transformation
- Bleach (Carl Roth, catalog number: 9062 )
- 37% HCl (Merck Millipore, catalog number: 100317 )
- Genotyping (plants)
- Optional: Phire Plant Direct PCR Kit (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: F-130WH )
- Optional: T7 endonuclease I (New England Biolabs, catalog number: M0302 ) plus NEBuffer 2 (New England Biolabs, catalog number: B7002 ), 250 mM EDTA
- Optional: pGEM®-T easy (Promega, catalog number: A3600)
- Oligonucleotides (5’-3’), 10 pmol/µl
- M13F_TGTAAAACGACGGCCAGT
- M13R_CAGGAAACAGCTATGACC
- K197 _CTGTTAATCAGAAAAACTCAG
Equipment
Note: No specific equipment is required. Any appropriate device can be used.
- Computer with internet access
- Thermocycler (e.g., Eppendorf, model: Mastercycler® nexus )
- Thermoblock (e.g., Eppendorf, model: Thermomixer® comfort )
- Plate incubators (28 °C, 37 °C)
- Shakers (28 °C, 37 °C)
- Horizontal gel-electrophoresis system (e.g., Bio-Rad Laboratories, model: Mini-Sub® Cell GT System )
- UV transilluminator (e.g., Bio-Rad Laboratories, model: GelDocTM XR+ System )
- Electroporator, (e.g., Bio-Rad Laboratories, model: MicroPulserTM Electroporator )
- 10 L desiccator
- Fume hood
- 200 ml beaker
Procedure
- Preparation of gRNA-spacers with CRISPR-PLANT© (Xie et al., 2014) and primer design (Figure 1I)
- Go to http://www.genome.arizona.edu/crispr/index.html.
- Select ‘Search’.
- Select ‘Select Species’ and choose ‘Arabidopsis thaliana’.
- Select ‘Chromosome’ and enter the coordinates of the target region in ‘From’ and ‘To’.
- Confirm with ‘Search by region’.
You will be returned Class 0.0 and Class 1.0 gRNAs. Class 0.0 gRNAs are supposed to be more specific. The SeqID is the coordinate of the gRNA in your predefined region. The minMM_GG (and minMM_AG) value refers to the minimum amount of mismatches the 12 bp sequence adjacent to the PAM motif has to any other sequence in the A. thaliana genome. The higher the value the smaller is the risk for off-targeting. Therefore, sequences with a minMM_GG higher than three should be considered. - Copy the optimal 20 bp long spacer sequence. Here, X represents any nucleotide.
Forward sequence: 5’-XXXXXXXXXXXXXXXXXXXX-3’
Reverse sequence: 5’-XXXXXXXXXXXXXXXXXXXX-3’
As the U6-promoter drives the expression of the gRNA, the initiation of transcription requires a guanine (G) as the transcription start site. If the chosen spacer sequence does not start with a ‘G’, change the first base. This will have no considerable effects on the specificity of Cas9 as the specificity largely depends on the 12 bp adjacent to the PAM motif.
Forward sequence: 5’-GXXXXXXXXXXXXXXXXXXX-3’
Reverse sequence: 5’-XXXXXXXXXXXXXXXXXXXC-3’ - Add ‘GATT’ and ‘AAAC’ to the 5’-ends of the forward and reverse sequences, respectively, to generate BpiI-compatible overhangs.
Forward primer: 5’-GATTGXXXXXXXXXXXXXXXXXXX-3’
Reverse primer: 5’-AAACXXXXXXXXXXXXXXXXXXXC-3’ - Now you have a complementary primer pair ready to order.
- Anneal the gRNA-primers (Figure 1II)
- Mix 10 µl of each primer (10 pmol/µl). Denature primers for 3 min at 98 °C. Hybridize primers in a thermocycler by decreasing the temperature from 98 °C to 22 °C. Pause at each degree for 30 sec.
- Digest the AtU6-26-V4-vector with BpiI according to manufacturer’s instructions (Figure 1III)
- Mix the digest with DNA-loading dye and load it on a 1% agarose gel. Separate digested from undigested bands by gel electrophoresis.
- Purify the linear vector backbone (3,517 bp) from gel.
- Ligate the annealed gRNA-primers and the BpiI-digested AtU6-26-V4-vector in a 10 µl ligation reaction (Figure 1IV).
- Mix 25 ng to 50 ng vector, 1 µl of 10x T4 DNA ligase reaction buffer and 0.5 µl T4 DNA ligase (200 U). Fill the rest of the volume with the hybridized primer.
- Incubate the reaction for 10 min at room temperature.
Note: Use 4 °C overnight incubation if you encounter problems getting positive transformants in step 6. - Use 3 µl of the ligation reaction to transform chemically competent TOP10 E. coli cells.
- Thaw 50 µl competent cells on ice.
- Add 3 µl of the ligation reaction to the cells.
- Incubate on ice for 15 min.
- Heat shock the cells for 45 sec at 42 °C in a thermoblock and place back on ice for 2 min.
- Add 400 µl of YT medium and incubate the cells in a thermoblock for 60 min at 37 °C and 750 rpm.
- Plate 60 to 80 µl of the transformation on solid YT medium with 50 µg/ml ampicillin (YT-amp).
- Do a colony-PCR to genotype transformants. Usually testing 5 to 10 colonies is sufficient to obtain positive transformants.
- Set up a 20 µl PCR reaction per colony as follows. Final concentrations are indicated in brackets:
2 µl 10x Taq buffer (1x)
0.6 µl M13F (0.5 µM)
0.6 µl gRNA-specific reverse primer (0.5 µM)
0.4 µl dNTPs (0.2 mM)
0.1 µl Green Taq DNA polymerase (0.5 U)
16.3 µl deionized water - Use a pipette tip to pick a colony and restreak the cells on an YT-amp plate. Directly afterwards dip the tip into the PCR reaction to transfer a small amount of cells. Incubate the plate at 37 °C for approximately 5 h and run the PCR in the thermocycler using the following conditions:

Note: The annealing temperature depends on the gRNA-specific reverse primer. M13F works well within the range of 54 °C to 60 °C during annealing. Instead of M13F and the gRNA-specific reverse primer you can also use M13R and the gRNA-specific forward primer with an elongation time of 20 sec. - Add 4 µl of 6x DNA loading dye to the reaction and load it on a 1% agarose gel. The expected amplicon size is 581 bp for the PCR reaction using M13F and the gRNA-specific reverse primer or 257 bp for the reaction with M13R and the gRNA-specific forward primer.
- Prepare 3 ml of YT-amp and pick the PCR-positive clones from the restreak plate. Incubate over night at 37 °C with 190 rpm.
- Isolate the plasmid from positive clones and check for the correct insert by Sanger sequencing using M13R as sequencing primer.
- Set up a 20 µl PCR reaction per colony as follows. Final concentrations are indicated in brackets:
- Optional (for multiplexing):
If more than one gRNA is needed, prepare each gRNA in a separate AtU6-26-V4-vector first (gRNA1-AtU6-26-V4, gRNA2-AtU6-26-V4, etc.) and combine them into one single gRNA2-gRNA1-AtU6-26-V4-vector.- Cut gRNA1-AtU6-26-V4 open with KpnI-HF and SpeI-HF (3,519 bp, Figure 1V).
- Cut gRNA2 out of gRNA2-AtU6-26-V4 using KpnI-HF and XbaI (643 bp, Figure 1VI).
- Load reactions on gel, gel purify positive bands.
- Ligate KpnI-gRNA2-XbaI and SpeI-gRNA1-AtU6-26-V4-KpnI (XbaI and SpeI produce compatible ends, Figure 1VII).
- Transform TOP10 E. coli cells, select transformants on YT-amp plates.
- Genotype clones with M13F and a specific reverse primer for the last added gRNA (amplicon size = 581 bp).
- Isolate plasmids and sequence with M13R.
Repeat this step to add more gRNAs.
- Cut gRNA1-AtU6-26-V4 open with KpnI-HF and SpeI-HF (3,519 bp, Figure 1V).
- Digest gRNA2-gRNA1-AtU6-26-V4 with KpnI-HF and SbfI-HF according to manufacturer’s instructions. With this step, you will cut out the gRNA2-gRNA1 unit including U6-promoters (Figure 1VIII).
- Mix the digest with DNA-loading dye and load it on a 1% agarose gel. Separate digested from undigested bands by gel electrophoresis.
- Purify the gRNA-fragment (660 bp for a single gRNA-fragment, 1,287 bp for a double gRNA-fragment) from gel.
- Mix the digest with DNA-loading dye and load it on a 1% agarose gel. Separate digested from undigested bands by gel electrophoresis.
- Digest UBQ10::pcoCas9p1300 with KpnI-HF and SbfI-HF according to manufacturer’s instructions to open the vector (Figure 1IX).
- Mix the digest with DNA-loading dye and load it onto a 1% agarose gel. Separate digested from undigested bands by gel electrophoresis.
- Purify the linearized vector backbone (14.3 kb) from gel.
- Mix the digest with DNA-loading dye and load it onto a 1% agarose gel. Separate digested from undigested bands by gel electrophoresis.
- Ligate the gRNA2-gRNA1 unit and UBQ10::pcoCas9p1300 in a 20 µl reaction (Figure 1X).
- Mix 25 ng to 50 ng vector, 10 ng to 20 ng gRNA2-gRNA1, respectively, 2 µl of 10x T4 DNA ligase reaction buffer, deionized water ad 19 µl, and 1 µl T4 DNA ligase (400 U).
- Incubate the reaction at 4 °C overnight.
- Mix 25 ng to 50 ng vector, 10 ng to 20 ng gRNA2-gRNA1, respectively, 2 µl of 10x T4 DNA ligase reaction buffer, deionized water ad 19 µl, and 1 µl T4 DNA ligase (400 U).
- Use 3 µl of the ligation reaction to transform chemically competent TOP10 E. coli cells. Select transformants on YT plates with 30 µg/ml kanamycin.
- Do a colony-PCR to genotype transformants as described in step 6.
- Genotype clones with K197 and a specific forward primer for the first added gRNA (here gRNA1, product size 801 bp).
- Isolate and sequence plasmids with M13F (optional with K197).
- Genotype clones with K197 and a specific forward primer for the first added gRNA (here gRNA1, product size 801 bp).
- Use 50 ng AtU6::gRNA2-AtU6::gRNA1-UBQ10::pcoCas9p1300 to transform A. tumefaciens via electroporation.
- Thaw 50 µl competent A. tumefaciens on ice.
- Pre-cool the cuvette on ice.
- Add 50 ng final vector to the A. tumefaciens cells, transfer to the cuvette and place the cuvette in the electroporator.
- Choose the ‘Agr’ settings at the MicroPulserTM electroporator (Bio-Rad, 1 pulse at 2.2 kV) or equivalent settings to transform the cells.
- Add 1 ml of YEB medium and transfer into a fresh 1.5 ml reaction tube. Incubate at 28 °C and 750 rpm for 2 to 3 h.
- Plate 60 µl of the transformation and select on YEB medium with 30 µg/ml kanamycin, 100 µg/ml rifampicin and 25 µg/ml gentamycin at 28 °C for 2 days.
- Thaw 50 µl competent A. tumefaciens on ice.
- Use the transformed A. tumefaciens to transform A. thaliana by floral dip with infiltration (Clough and Bent, 1998; Chen, 2011)
- Sterilize seeds, e.g., by vapor-phase sterilization.
- Transfer seeds to 1.5 ml reaction tubes.
- Place open tubes in a 10 L desiccator under the fume hood.
- Place a 200 ml beaker filled with 100 ml bleach (6%) into the desiccator.
- Use a glass pipette to add 3 ml HCl (37%) to the bleach and close desiccator lid quickly.
Note: Careful: The vapor can chemically burn skin and eyes! - Sterilize for 3 to 4 h.
- Transfer seeds to 1.5 ml reaction tubes.
- Sow seeds on ½ MS with 20 µg/ml hygromycin or kanamycin (depending on the resistance marker from the UBQ10::pcoCas9p1300 of your choice) to select for positive transformants. Seal plates with MicroporeTM tape. Stratify seeds in darkness at 4 °C for 3 days. Germinate seeds in long day conditions (22 °C, 16:8 h light:dark photoperiod) until transformants reach 4 true-leaf stage. Transplant seedlings to soil and continue growth in long day conditions.
- Genotype the transformants.
As in the T1 generation the activity and efficiency of the Cas9 enzyme but also of the DNA repair mechanisms might differ from cell to cell, each plant most likely will be a mosaic of different genotypes. Therefore we suggest to pooling genotyping samples of one plant, such as pieces of cauline leaves from different branches. If a plant with the expected mutation was identified in T1, label and genotype the single branches as they can have different genotypes (Figures 2C and 2D). The method of genotyping depends on the anticipated results. - Big fragment deletions which had been generated by multiple gRNAs, can be identified by PCR followed by gel electrophoresis of the PCR products (Figures 2A and 2B).
Note: Design primers flanking the expected deletion in a distance of approximately 200 bp to 400 bp to each gRNA target site. For high throughput genotyping we suggest to prepare 10 µl PCR reactions using the Phire Plant Direct PCR Kit.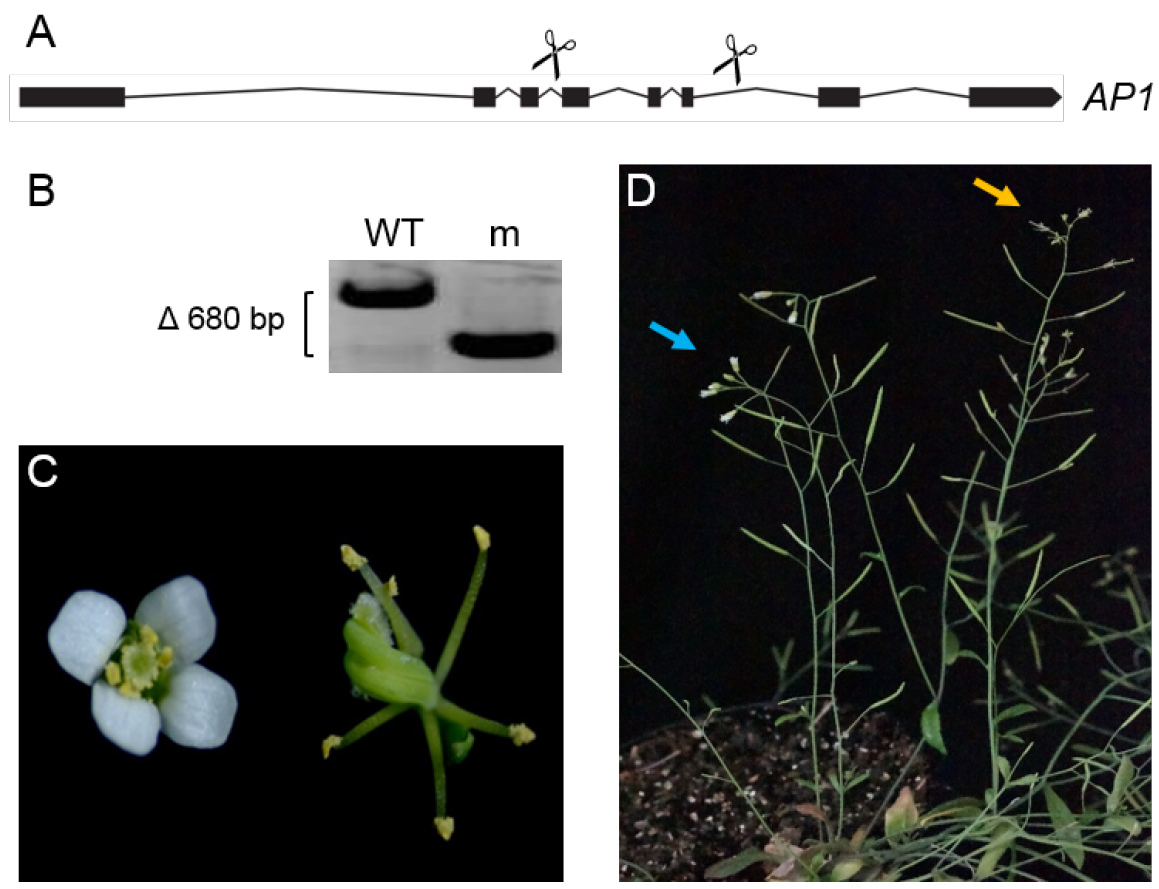
Figure 2. Detection of big fragment deletions in the AP1 locus. A. The AP1 locus was targeted using two different gRNAs (scissors). Target sites are sequences in the third and sixth intron of AP1. B. Genotyping of a phenotypically wild type (WT) and an ap1-like (m) branch in the T1 generation revealed the presence of a 680 bp deletion in the AP1 locus in the ap1-like branch (m). C and D. Plants in the T1 generation can be a mosaic of different genotypes. Two different flower phenotypes, wild type (C, left; D, blue arrow) and ap1 (C, right; D, yellow arrow) can be found on two different branches of the same plant. - Small deletions or polymorphisms that produce a minimum mismatch of 2 bp to the wild type allele can be detected by T7 endonuclease digestion (Vouillot et al., 2015).
- Perform the genotyping PCR as mentioned for big fragment deletions.
- Set up the hybridization reaction by adding 1.12 µl NEBuffer 2 to the PCR product without purification of the DNA. Prepare WT control and sample separately. When pooling different branches in T1, it is not necessary to add wild type PCR product to the sample as we usually do not expect Cas9 efficiencies to be higher than the T7 endonuclease detection limit (Vouillot et al., 2015). But if you already see a clear phenotype when genotyping single branches, you can replace 20% of the sample volume by wild type to not miss homozygous mutant branches.
- Hybridize PCR products using the following conditions:
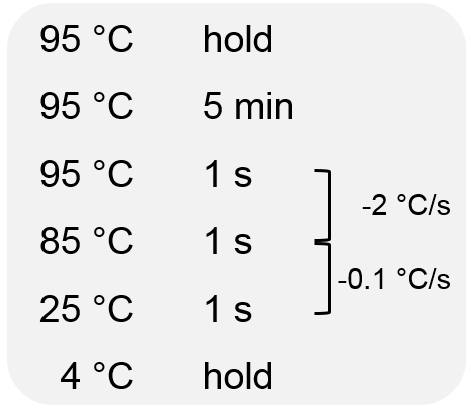
- Add 0.1 µl of T7 endonuclease (1 U) and incubate for 30 min at 37 °C.
- Stop the reaction with 0.8 µl 250 mM EDTA.
- Separate cleavage products on a 2% agarose gel.
- Perform the genotyping PCR as mentioned for big fragment deletions.
- Point mutations are not detected by T7 endonuclease digest efficiently (Vouillot et al., 2015) and the IDT® Surveyor® nuclease (Integrated DNA Technologies), which is supposed to cut at point mutations, appeared to be incompatible with the Phire Plant Direct PCR Mix. Considering the economic factors of high throughput mutant screening, we did not increase the amount of Surveyor® enzyme beyond manufacturer’s recommendations. Here, we suggest to using the T7 endonuclease assay to exclude plants with mutations of 2 bp or more. Then PCR products from the rest of the samples can be purified and sent for Sanger sequencing.
- Confirm the mutation by sequencing.
- Harvest the seeds from the branch that has a mutation.
- Sow the seeds to generate a small T2 population of approximately 100 individuals. Select the plant that carries the mutation but lost the T-DNA.
- To identify plants that still carry the transgene, use M13F and the gRNA-specific reverse primer (product size 557 bp). Exclude PCR-positive plants.
- Genotype for the expected mutation as described in step 17.
Note: To verify small fragment deletions in T2 (Figure 3), prepare WT, sample and a 1:1 mix of WT and sample prior to hybridization. The WT-sample-mix is necessary as plants can be homozygous for the mutant allele. In that case, they do not produce heteroduplex DNA when hybridized and will not be digested by T7 endonuclease.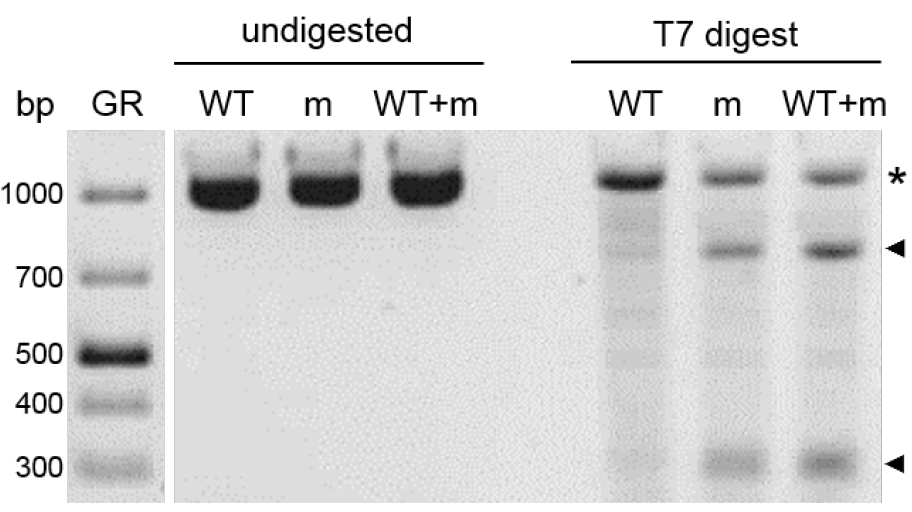
Figure 3. Detection of small deletions or polymorphisms by T7 digest. PCR samples of T2 wild type (WT), mutant (m) and a 1:1 mix of wild type and mutant (WT+m) were hybridized. Half of the reaction volume was incubated without T7 endonuclease (undigested) and the other half was incubated with 1 U of the enzyme (T7 digest) and separated by gel electrophoresis on 2% agarose. The presence of cleavage products in the mutant sample is the result of at least two different alleles, which can be wild type and mutant or different mutant alleles or a mix of both options. As the addition of wild type to the mutant sample (WT+m) does not dilute the signal of the cleavage products, there are most likely different mutant alleles, which later was confirmed by sequencing (not shown). Asterisk indicates undigested PCR products, black arrows point out cleavage products. GR indicates the GeneRulerTM 1 kb Plus DNA ladder.
Note: Once the protocol is set up, the ‘undigested’ controls can be omitted.
Data analysis
Mutations obtained in the T1 generation can be confirmed by Sanger sequencing as mentioned in step 17. But, as neighboring cells might harbor different mutations and as the mutation might be biallelic, we recommend to gel-purify the mutant PCR product and subclone it to pGEM®-T or any other cloning vectors. Sequence five to ten positive colonies to get an overview of gained mutations. After the selection of a T-DNA-free mutant plant in the T2 generation (step 19) confirm the presence and type of the mutation again by sequencing.
Note: PCR products generated by the Phire Plant Direct PCR Kit are blunt ended and require A-tailing before ligation with pGEM®-T.
Notes
This protocol is simple and straightforward. So far, more than 50 binary vectors containing different sets of gRNAs have been generated by our group and used for genome editing of Arabidopsis.
Recipes
- 10x TBE electrophoresis buffer (1 L)
1 M Tris
1 M boric acid
0.02 M EDTA
Deionized water ad 1,000 ml
Dilute 10x TBE buffer with deionized water to 1:10 to use as gel running buffer - YEB medium for Agrobacterium (1 L)
5 g beef extract
1 g yeast extract
5 g peptone
5 g sucrose
500 mg MgSO4
10 g Bacto-agar
Deionized water ad 1,000 ml
Adjust pH to 7.0 using NaOH
Autoclave YEB medium at 121 °C and 2 bar for 15 min - YT medium for E. coli (1 L)
5 g NaCl
5 g yeast extract
8 g peptone
(15 g Bacto-agar)
Deionized water ad 1,000 ml
Adjust pH to 7.0 using NaOH
Autoclave YT medium at 121 °C and 2 bar for 15 min - ½ MS for Arabidopsis (1 L)
2.3 g MS + B5 Vitamins
8 g Phyto-agar
Deionized water ad 1,000 ml
Adjust pH to 5.7 using KOH
Autoclave ½ MS at 121 °C and 2 bar for 15 min
Prepare sterile 50% sucrose (filter sterilize or autoclave separately)
Add 1% sucrose to ½ MS prior to pouring the plates
Note: The addition of sucrose is not required. The advantage is that seedlings recover better on selective media. The disadvantage is that it promotes fungal growth on the medium.
Acknowledgments
This protocol is based on our work previously published in Plant Methods (Yan et al., 2016). Work of J.S. was supported by the IMPRS-PMPG fellowship. K.K. wishes to thank the Alexander-von-Humboldt foundation and the BMBF for support. The authors declare that they have no competing interests.
References
- Chen, L. (2011). Simplified Agrobacterium-mediated transformation of Arabidopsis thaliana. Bio-protocol Bio101: e41.
- Clough, S. J. and Bent, A. F. (1998). Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16(6): 735-743.
- Li, J. F., Norville, J. E., Aach, J., McCormack, M., Zhang, D., Bush, J., Church, G. M. and Sheen, J. (2013). Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat Biotechnol 31(8): 688-691.
- Mersereau, M., Pazour, G. J. and Das, A. (1990). Efficient transformation of Agrobacterium tumefaciens by electroporation. Gene 90(1):149-51.
- Nekrasov, V., Staskawicz, B., Weigel, D., Jones, J. D. and Kamoun, S. (2013). Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat Biotechnol 31(8): 691-693.
- Shan, Q., Wang, Y., Li, J., Zhang, Y., Chen, K., Liang, Z., Zhang, K., Liu, J., Xi, J. J., Qiu, J. L. and Gao, C. (2013). Targeted genome modification of crop plants using a CRISPR-Cas system. Nat Biotechnol 31(8): 686-688.
- Sorek, R., Lawrence, C. M. and Wiedenheft, B. (2013). CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu Rev Biochem 82: 237-266.
- Vouillot, L., Thelie, A. and Pollet, N. (2015). Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered by engineered nucleases. G3 (Bethesda) 5(3): 407-415.
- Xie, K., Zhang, J. and Yang, Y. (2014). Genome-wide prediction of highly specific guide RNA spacers for CRISPR-Cas9-mediated genome editing in model plants and major crops. Mol Plant 7(5): 923-926.
- Yan, W., Chen, D. and Kaufmann, K. (2016). Efficient multiplex mutagenesis by RNA-guided Cas9 and its use in the characterization of regulatory elements in the AGAMOUS gene. Plant Methods 12: 23.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Schumacher, J., Kaufmann, K. and Yan, W. (2017). Multiplexed GuideRNA-expression to Efficiently Mutagenize Multiple Loci in Arabidopsis by CRISPR-Cas9. Bio-protocol 7(5): e2166. DOI: 10.21769/BioProtoc.2166.
Category
Plant Science > Plant transformation > Agrobacterium
Plant Science > Plant developmental biology > General
Molecular Biology > DNA > DNA modification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.