- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Efficient AAV-mediated Gene Targeting Using 2A-based Promoter-trap System

Published: Vol 6, Iss 24, Dec 20, 2016 DOI: 10.21769/BioProtoc.2058 Views: 10868
Reviewed by: Nicoletta CordaniPinchas TsukermanMartin V Kolev
Original research article
The authors used this protocol in:

Apr 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Adeno-associated virus (AAV)-based targeting vectors have 1-4-log higher gene targeting efficiencies compared with plasmid-based targeting vectors. The efficiency of AAV-mediated gene targeting is further increased by introducing a promoter-trap system into targeting vectors. In addition, we found that the use of ribosome-skipping 2A peptide rather than commonly used internal ribosome entry site (IRES) in the promoter-trap system results in significantly higher AAV-mediated gene targeting efficiencies (Karnan et al., 2016). In this protocol, we describe the procedures for AAV-mediated gene targeting exploiting 2A for promoter trapping, including the construction of a targeting vector based on the platform plasmid pAAV-2Aneo or pAAV-2Aneo v2, production of AAV particles, infection of cells with resulting AAV-based targeting vectors, and isolation and verification of gene-targeted cell clones.
Keywords: Adeno-associated virusBackground
The procedures for AAV-mediated gene targeting in general (corresponding to Sections B-G of this protocol) were previously described in other protocols (Kohli et al., 2004; Rago et al., 2007; Khan et al., 2011; Howes and Schofield, 2015). However, this protocol provides a detailed description of how to perform AAV-mediated gene targeting using a 2A-based promoter–trap system for the first time.
Materials and Reagents
- Pipette tips
- 10-cm culture dish
- 1.5-ml or 2-ml cryovial
- 96-well plate
- 15-ml (or 50-ml) conical tube
- 24-well plates
- 6-well plates or tissue culture dishes/flasks
- 0.22 µm filter
- 1.5-ml tubes
- Aluminum foil
- Disposable pipetting reservoir (AS ONE, catalog number: 2-7844-02 )
- E. coli DH5α competent cells (Takara Bio, catalog number: 9057 )
Note: This product has not been discontinued. - HEK293 or HEK293T cell line
- Cell line(s) for gene targeting
- pAAV-2Aneo (Addgene, catalog number: 80032 )
- pAAV-2Aneo v2 (Addgene, catalog number: 80033 )
- Agarose (NIPPON GENE, catalog number: 318-01195 )
- Pwo SuperYield DNA polymerase (Roche Diagnostics, catalog number: 04340850001 )
- PureLink® Quick Gel Extraction Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: K2100-25 )
- PureLink® PCR Purification Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: K3100-02 )
- Quick LigationTM Kit (New England BioLabs, catalog number: M2200S )
- Dry ice
- Restriction enzymes BspEI, MluI, or BsrGI (New England BioLabs, Ipswich, MA)
- Restriction enzyme buffers
- LB broth (NACALAI TESQUE, catalog number: 20068-75 )
- Mini PlusTM Plasmid DNA extraction system (Viogene, catalog number: GF2002 )
- BigDye® Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 4337455 )
- Alkaline phosphatase, calf intestinal (CIP) (New England BioLabs, catalog number: M0290L )
- Custom-made PCR primers (for the amplification of 5’ and 3’ homology arms)
- Custom-made sequencing primers (for the sequencing of 5’ and 3’ homology arms)
- Custom-made PCR primers (for the screening of gene-targeted clones)
- PureLink® HiPure Plasmid Maxiprep Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: K210007 )
- Dulbecco’s modified Eagle’s medium (D-MEM) (Wako Pure Chemical Industries, catalog number: 044-29765 )
- Fetal bovine serum (FBS) (NICHIREI, catalog number: 172012-500ml )
- Opti-MEM® I reduced-serum medium (Thermo Fisher Scientific, GibcoTM, catalog number: 31985-070 )
- pRC and pHelper plasmids in AAV Helper-free system (Agilent Technologies, catalog number: 240071 )
- TransIT®-293 transfection reagent (Mirus Bio, catalog number: MIR 2705 )
- Penicillin-streptomycin solution (x100) (Wako Pure Chemical Industries, catalog number: 168-23191 )
- Methanol
- Distilled water
- RNase-free DNase set (QIAGEN, catalog number: 79254 )
- NeoR-Rev #1: 5’-GGCATCAGAGCAGCCGATTG
- NeoR-Fwd #1: 5’-CATTCGACCACCAAGCGAAA
- NeoR-Rev #2: 5’-CTTGAGCCTGGCGAACAGTT
- KOD FX Neo (TOYOBO, catalog number: KFX-201 )
- Growth medium appropriate for the cell line(s) undergoing gene targeting
- 0.25% (w/v) trypsin solution with phenol red (Wako Pure Chemical Industries, catalog number: 201-18841 )
- PCR buffer
- dNTPs
- SYBR® Green I nucleic acid stain (Lonza, catalog number: 50512 )
- PureLink® Genomic DNA Mini Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: K182002 )
- Ampicillin sodium (Wako Pure Chemical Industries, catalog number: 012-23303 )
- Dimethyl sulfoxide (DMSO) (Wako Pure Chemical Industries, catalog number: 048-21985 )
- NaCl
- KCl
- Na2HPO4•12H2O
- KH2PO4
- Ampicillin working solution (see Recipes)
- SYBR Green I working solution (see Recipes)
- PBS(-) (see Recipes)
Equipment
- Humidified CO2 incubator
- VeritiTM thermal cycler (Thermo Fisher Scientific, model: Veriti Thermal Cycler )
- Mupid®-2plus submarine-type electrophoresis system (Takara Bio, model: Mupid-2plus System )
- NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific)
- StepOnePlusTM Real-Time PCR system (Thermo Fisher Scientific, Applied BiosystemTM, model: StepOnePlusTM Real-Time PCR system )
- Centrifuge for molecular biology experiments
- -20 °C freezer
- Tissue culture hood
- Phase-contrast microscope
- Swing bucket centrifuge for tissue culture
- -80 °C deep freezer
- Water bath
- 12-channel pipettor
- Two permanent markers of different colors
Procedure
- Construction of an AAV-based targeting vector
- Design the PCR primers used to amplify homology arms. To incorporate the amplified homology arms into pAAV-2Aneo (v2) via restriction enzyme digestion, select appropriate restriction enzyme sites from the lineup of the multiple cloning sites MCS-1 (between SacI and SpeI sites) and MCS-2 (between KpnI and XhoI sites) in pAAV-2Aneo (v2). The selected restriction enzyme sites should be absent within the homology arms to be amplified. Then, introduce the selected restriction enzyme sites at the 5’ end of PCR primers. In addition, append 3-6 nucleotides (any sequence; the length depends on the restriction enzyme sites selected) distal to the restriction enzyme sites, which allows the enzymes to cleave PCR products close to the ends. For tips on designing homology arms, refer to the Notes section (Notes 1-3).
- Amplify the 5’ and 3’ homology arms by PCR. Use a high-fidelity DNA polymerase; e.g., Pwo SuperYield DNA polymerase.
- Analyze the PCR products by agarose gel electrophoresis. Cut out a band of the expected size from the gel, and recover a DNA fragment using the PureLink Quick Gel Extraction Kit. If the PCR product appears as a clean single band and a primer dimer is not detected on the agarose gel, the PCR product may optionally be purified using the PureLink PCR Purification Kit instead of performing gel extraction.
- Cleave the PCR products with the selected restriction enzymes, and purify the reaction products using the PureLink PCR Purification Kit.
- Since pAAV-2Aneo and pAAV-2Aneo v2 have different ‘frame adjusters’, these vectors are processed in different fashions to adjust reading frames. Follow one of the three options below (steps A5a-c) to process the frame adjuster. In any of the three options, the restriction enzyme-digested DNA solution should be purified using the PureLink PCR Purification Kit or other methods before self-religation. Self-religation is performed using the Quick Ligation Kit.
- When pAAV-2Aneo is used and the promoter-trap module is to be incorporated into an intron upon gene targeting (Figure 1): Cleave pAAV-2Aneo with one of the three restriction enzymes (BspEI, MluI, or BsrGI), and religate the vector so that 2Aneo is translated in frame with the 5’ portion of the endogenous target gene (Figure 2).
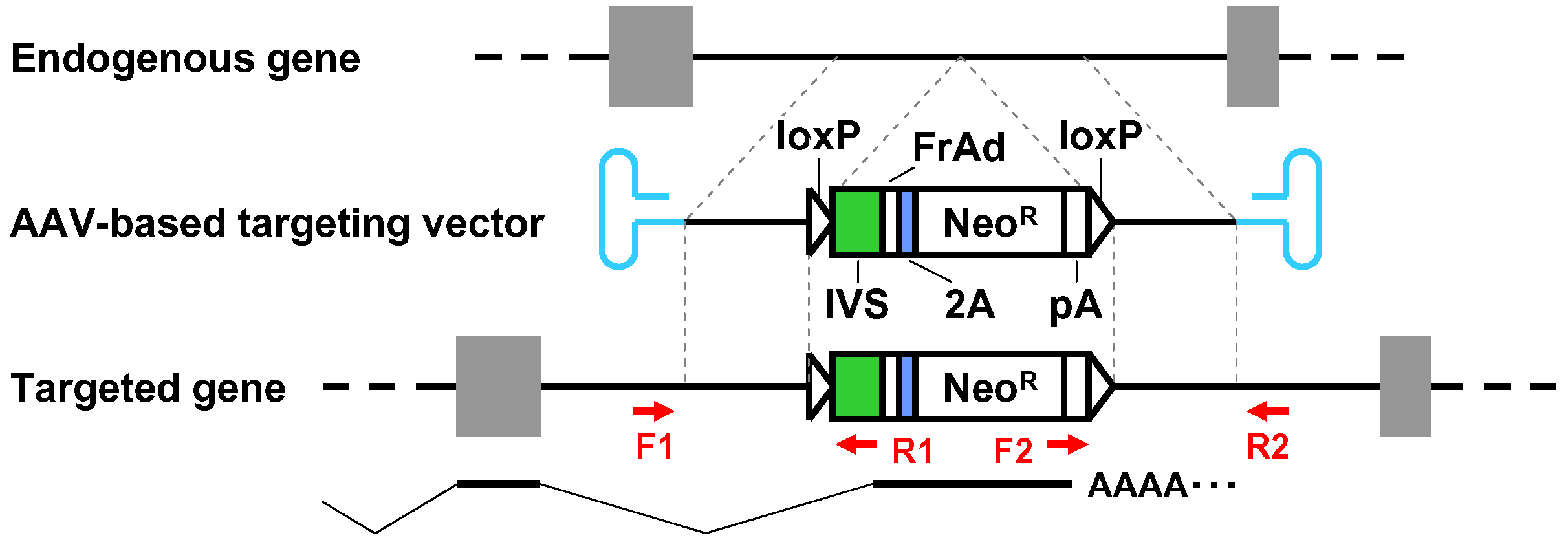
Figure 1. Schematic diagram of the AAV-mediated targeting of an intron. Thin dotted lines indicate homology between the endogenous genomic locus and the targeting vector. F1-R1 and F2-R2 indicate pairs of oligonucleotide primers used for screening PCR and confirmatory PCR. The bottom part shows a predicted mRNA expressed from the targeted genomic locus. Gray rectangles: exons; bold black lines: introns; blue hairpin structures: inverted terminal repeats; IVS: synthetic intron; FrAd: frame adjuster; NeoR: the neomycin phosphotransferase gene; pA: polyadenylation site.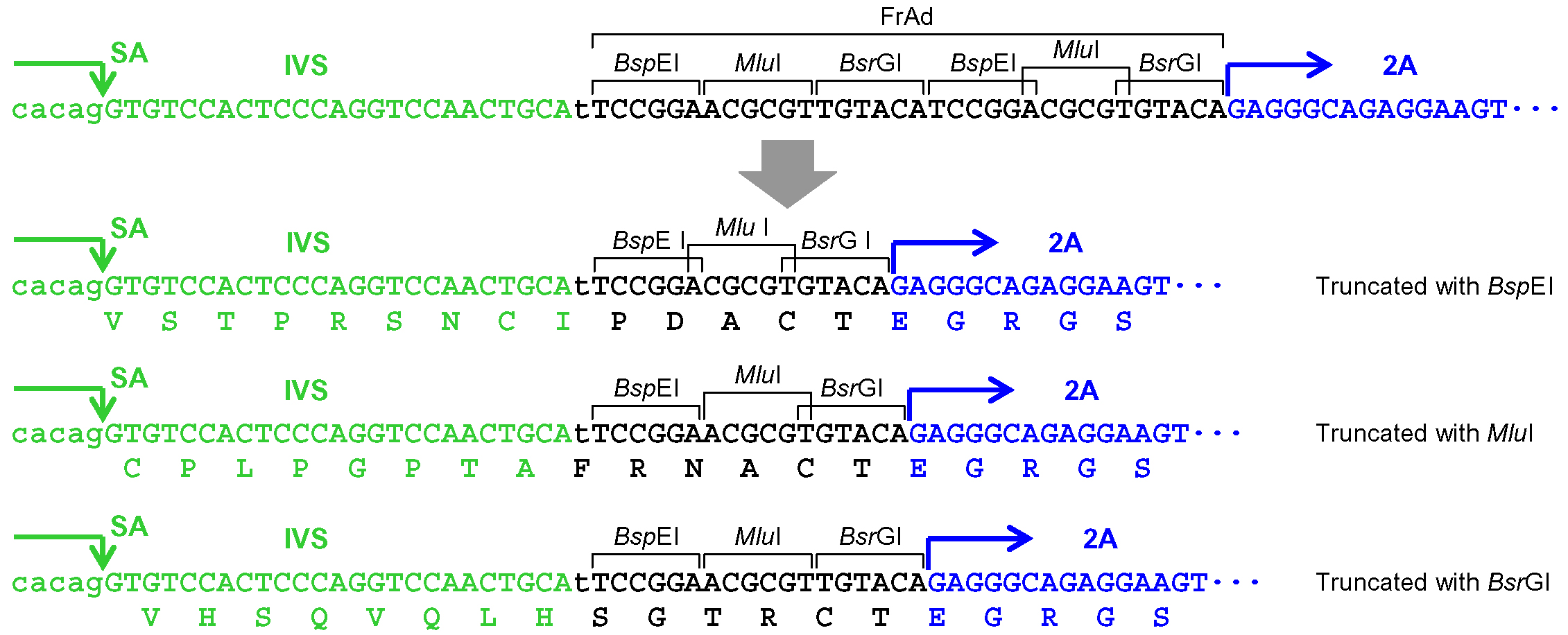
Figure 2. Processing of the frame adjuster in pAAV-2Aneo for targeting introns. An intact frame adjuster (top) and the ones processed by BspEI, MluI, and BsrGI digestion (bottom). Predicted partial polypeptides translated from the region surrounding the frame adjuster are shown under nucleotide sequences. SA: splice acceptor; IVS: synthetic intron; FrAd: frame adjuster. - When the promoter-trap module is to be incorporated into an intron upon gene targeting similar to step A5a (Figure 1), but pAAV-2Aneo v2 is used instead of pAAV-2Aneo: Cleave pAAV-2Aneo v2 with either BspEI or BsrGI and religate the vector, or leave the vector unprocessed, so that 2Aneo is translated in frame with the 5’ portion of the endogenous target gene (Figure 3). Figure 4 schematically depicts the disruption of PIGA intron 5 performed in our previous study as an example of option A5b.
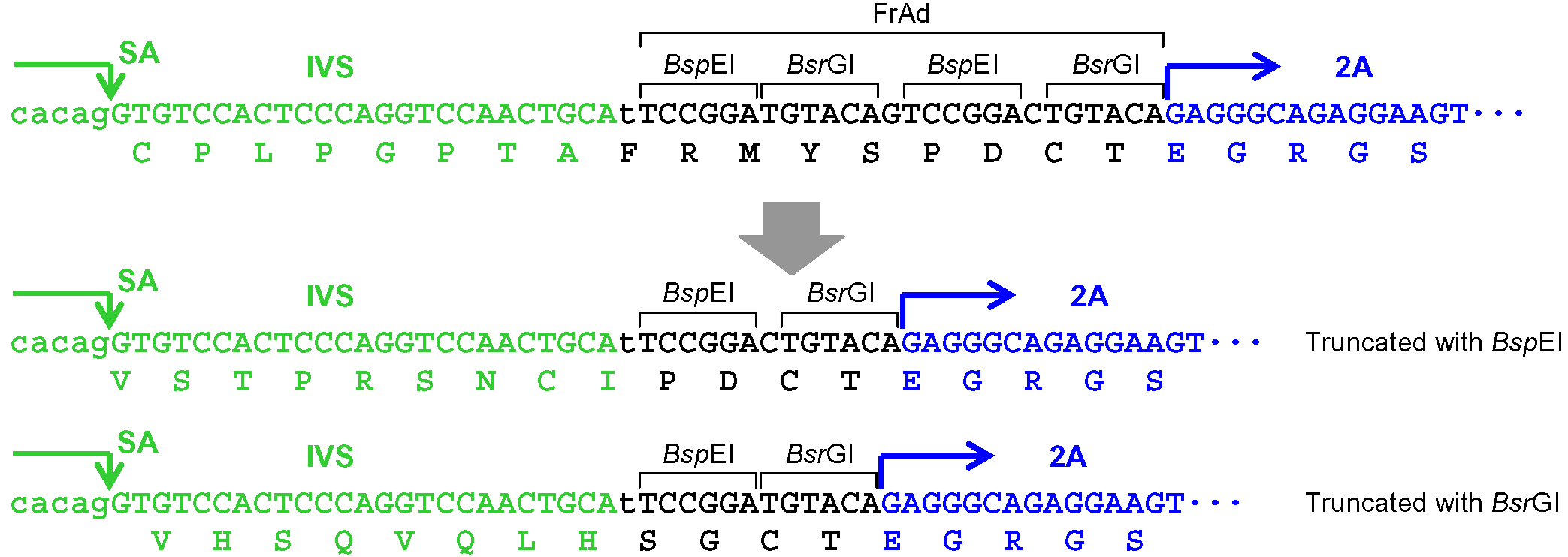
Figure 3. Processing of the frame adjuster in pAAV-2Aneo v2 for targeting introns. An intact frame adjuster (top) and the ones processed by BspEI and BsrGI digestion (bottom). Predicted partial polypeptides translated from the region surrounding the frame adjuster are shown under nucleotide sequences. SA: splice acceptor; IVS: synthetic intron; FrAd: frame adjuster.
Figure 4. Schematic description of the disruption of PIGA intron 5. Depicted at the top is the PIGA gene along with the nucleotide sequence of exon 5-intron 5 boundary and a corresponding partial PIGA protein. Shown at the bottom is a partial nucleotide sequence of the targeting vector including a BspE1-truncated frame adjuster. The amino acid sequence under the nucleotide sequence indicates a predicted translation product derived from the targeting vector. IVS: synthetic intron; FrAd: frame adjuster; NeoR: the neomycin phosphotransferase gene; pA: polyadenylation site; SD: splice donor; SA: splice acceptor. - When the promoter-trap module is incorporated into an exon, regardless of the vector platform employed (pAAV-2Aneo or pAAV-2Aneo v2; Figure 5): Cleave pAAV-2Aneo (v2) with BsrGI, and religate the vector so that 2Aneo is translated in frame with the 5’ portion of the endogenous target gene (Figures 6 and 7).
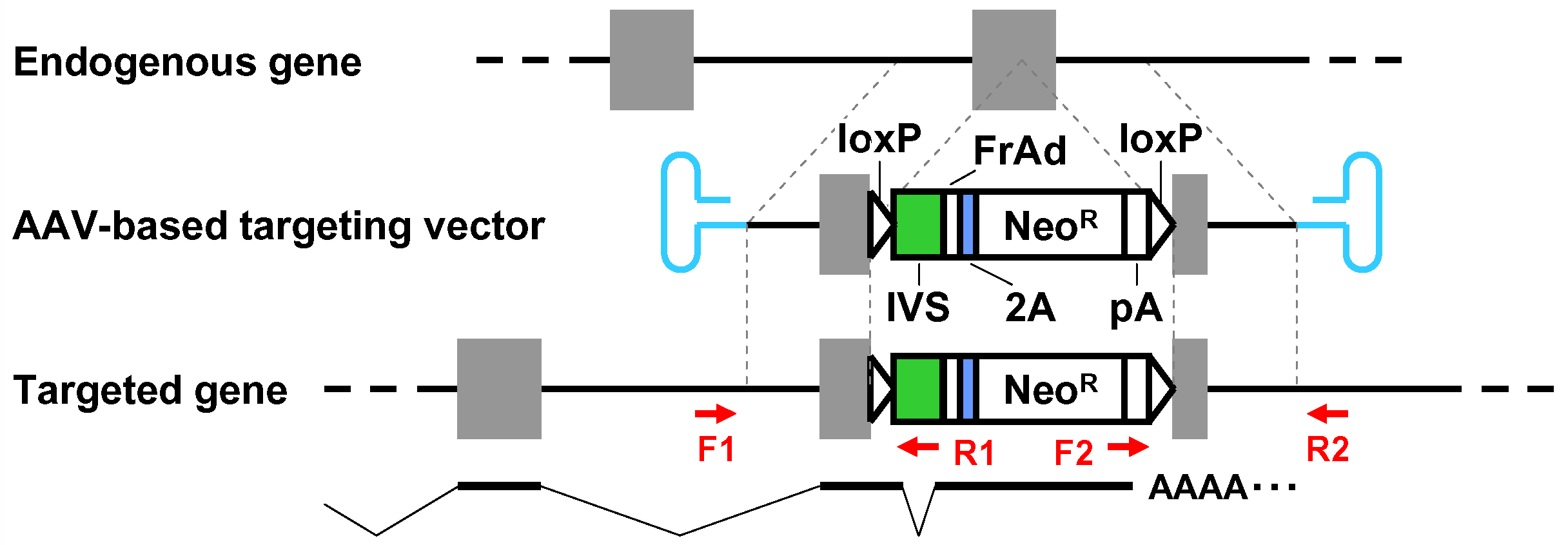
Figure 5. Schematic diagram of the AAV-mediated targeting of an exon. See the legend for Figure 1 for descriptions and abbreviations.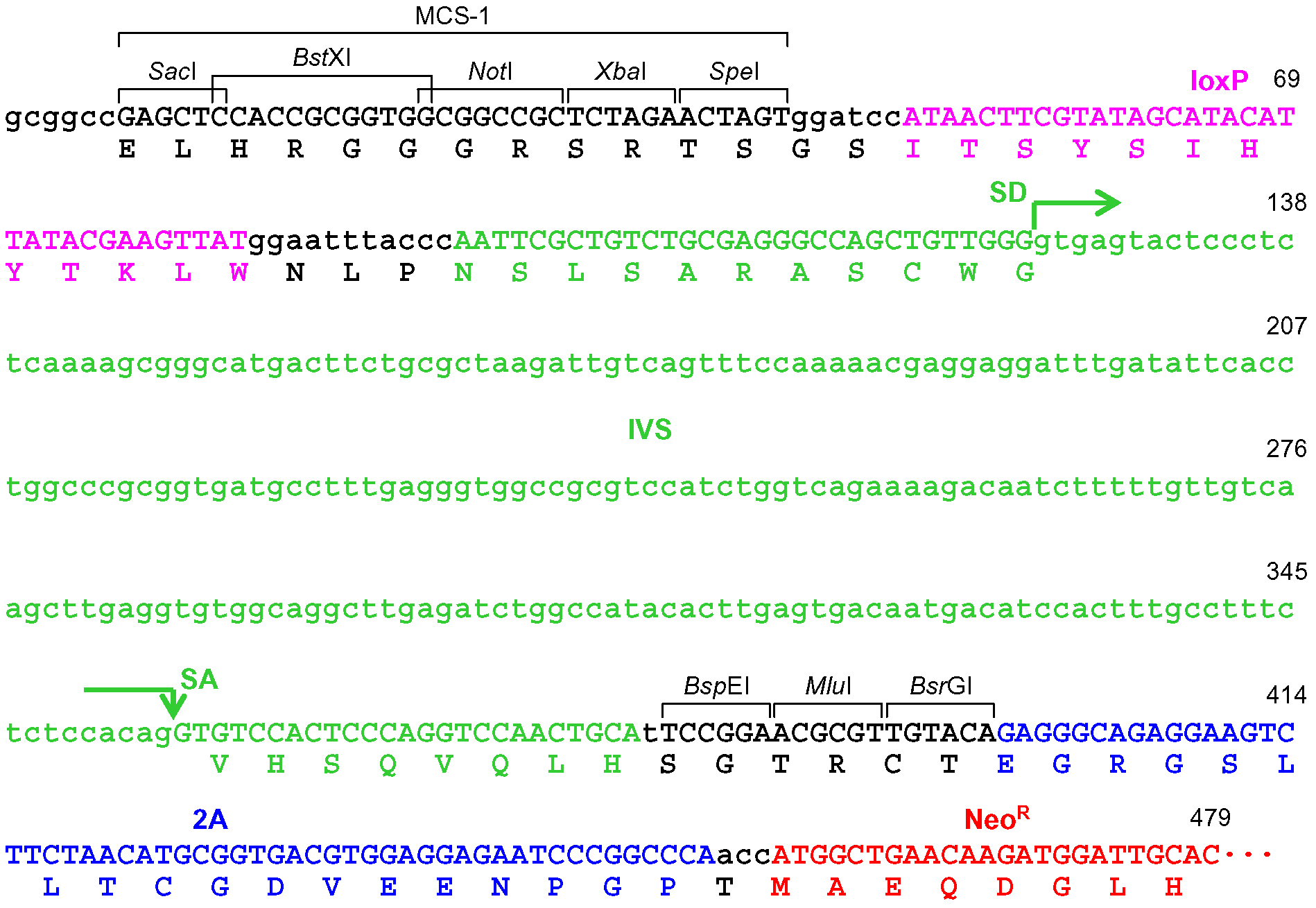
Figure 6. The frame adjuster truncated with BsrGI for exon targeting and its adjacent region in pAAV-2Aneo. A predicted partial polypeptide translated from this region is indicated under the nucleotide sequence. The 5’ homology arm should be designed such that the coding exon to be disrupted is translated in frame with the indicated polypeptide. See the legend for Figure 4 for abbreviations.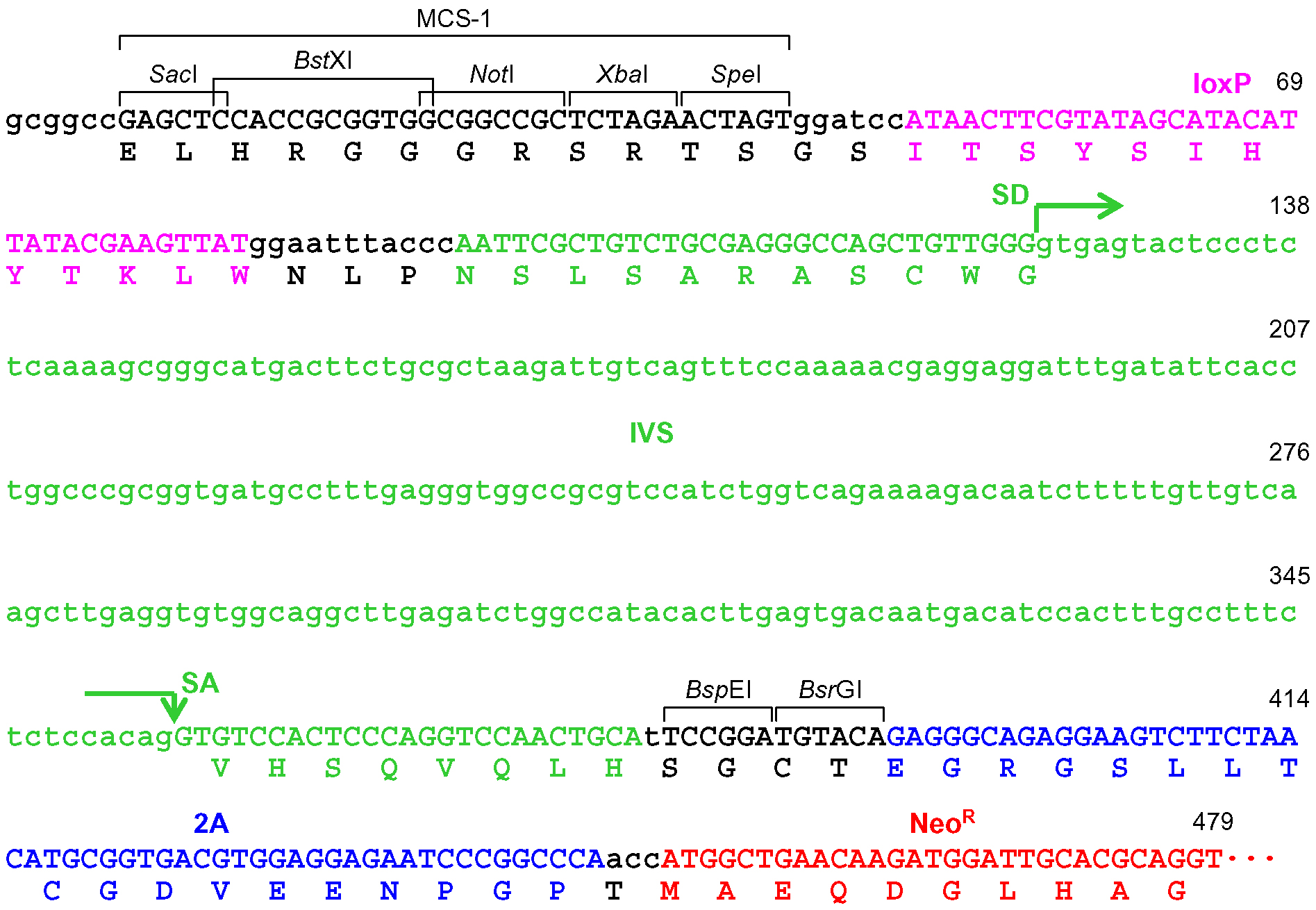
Figure 7. The frame adjuster truncated with BsrGI for exon targeting and its adjacent region in pAAV-2Aneo v2. See the legend for Figure 6 for descriptions and abbreviations.
- When pAAV-2Aneo is used and the promoter-trap module is to be incorporated into an intron upon gene targeting (Figure 1): Cleave pAAV-2Aneo with one of the three restriction enzymes (BspEI, MluI, or BsrGI), and religate the vector so that 2Aneo is translated in frame with the 5’ portion of the endogenous target gene (Figure 2).
- Transform competent cells with the ligation product following standard procedures.
- Spread the transformed competent cells onto an LB plate containing 50 µg/ml of ampicillin.
- Incubate the cells overnight at 37 °C.
- Pick several colonies with pipette tips, and culture the cells in 2.5 ml of LB containing 50 µg/ml of ampicillin, overnight at 37 °C.
- Perform mini-prep using the Mini Plus plasmid DNA extraction system as per manufacturer’s instructions.
- Confirm that the frame adjuster has been truncated as intended by sequencing the recovered plasmids using the BigDye Terminator v3.1 Cycle Sequencing Kit and a sequencing primer (e.g., NeoR-Rev #1, a primer complementary to the NeoR gene).
- To clone one of the PCR-amplified homology arms, cleave the plasmid verified in step A11 with an appropriate restriction enzyme(s). If required, use alkaline phosphatase to prevent self-religation of the plasmid. Purify the linearized plasmid using the PureLink PCR Purification Kit.
- Ligate the linearized plasmid with the PCR-amplified homology arm that was trimmed and purified in step A4.
- Repeat steps A6-A10 with the ligation product.
- Check for the successful insertion of the homology arm by digesting the resultant plasmids (mini-preps) with an appropriate restriction enzyme(s) and resolving the DNA fragments on an agarose gel.
- Verify the sequence of the PCR-amplified homology arm using the BigDye Terminator v3.1 Cycle Sequencing Kit and custom-made primers.
Note: The homology arm is incorporated between an inverted terminal repeat (ITR) and a loxP site, both of which are located close to MCS. Since both ITR and loxP are partially palindromic and thus (particularly ITR) may hinder DNA polymerization, we designed custom-made sequencing primers within the homology arms. - To clone another homology arm, cleave the plasmid from step A16 using an appropriate restriction enzyme(s) with or without alkaline phosphatase, and purify the linearized plasmid using the PureLink PCR purification kit. Then, repeat steps A13-A16.
- Perform maxi-prep of the constructed AAV-based targeting plasmid using the PureLink HiPure Plasmid Maxiprep Kit.
- Determine the concentration of the maxi-prep plasmid DNA with a NanoDrop spectrophotometer.
- Design the PCR primers used to amplify homology arms. To incorporate the amplified homology arms into pAAV-2Aneo (v2) via restriction enzyme digestion, select appropriate restriction enzyme sites from the lineup of the multiple cloning sites MCS-1 (between SacI and SpeI sites) and MCS-2 (between KpnI and XhoI sites) in pAAV-2Aneo (v2). The selected restriction enzyme sites should be absent within the homology arms to be amplified. Then, introduce the selected restriction enzyme sites at the 5’ end of PCR primers. In addition, append 3-6 nucleotides (any sequence; the length depends on the restriction enzyme sites selected) distal to the restriction enzyme sites, which allows the enzymes to cleave PCR products close to the ends. For tips on designing homology arms, refer to the Notes section (Notes 1-3).
- AAV production
- Plate HEK293 or HEK293T cells onto a 10-cm culture dish at an approximate density of 1 x 106 cells/dish with 10 ml of penicillin/streptomycin-free D-MEM supplemented with 5% FBS. The cells should be 70-80% confluent on the following day.
- On the following day, aspirate the medium, and add 5 ml of Opti-MEM I reduced-serum medium.
Note: Washing the cells prior to the addition of Opti-MEM I reduced-serum medium is optional; however, proceed with caution when washing, as the HEK293(T) cells are loosely adherent to the culture dish. - Pipette the following plasmids into 1.5 ml of Opti-MEM I reduced-serum medium and gently mix:
pRC 5 µg
pHelper 5 µg
AAV-based targeting plasmid 5 µg - Add 45 μl of TransIT-293 reagent, and gently mix by pipetting.
- Let the mixture stand for 15 min at room temperature.
- Pipette the DNA-reagent mixture drop-wise onto the HEK293(T) cells in the 10-cm culture dish, and gently mix by rocking the dish.
- Incubate the cells in a CO2 incubator at 37 °C for 24 h.
Notes:- A total of 6.5 ml medium is sufficient for 24 h incubation of cells in a 10-cm culture dish.
- The CO2 incubator is used at 37 °C and 5% CO2 under humidified conditions throughout the study.
- After 24 h of transfection, add 5 ml of D-MEM supplemented with 5% FBS and 1% penicillin/streptomycin.
- Plate HEK293 or HEK293T cells onto a 10-cm culture dish at an approximate density of 1 x 106 cells/dish with 10 ml of penicillin/streptomycin-free D-MEM supplemented with 5% FBS. The cells should be 70-80% confluent on the following day.
- Harvest of AAV particles
- After 72 h of transfection, scrape HEK293(T) cells from the 10-cm culture dish by extensive pipetting, and transfer the cells to a 15-ml conical tube with their supernatant.
- Centrifuge the 15-ml tube at 400 x g (1,500 rpm) for 5 min at room temperature, and discard the supernatant completely but carefully so as not to remove the cells.
- Wash the cells with PBS(-), centrifuge the 15-ml tube at 400 x g for 5 min, and discard the PBS(-) completely but carefully so as not to remove the cells.
- Resuspend the cell pellet in 1.0 ml of PBS(-).
Note: If the cell line that will be infected with the AAV vector in preparation is already known, the cell pellet in step C4 can be resuspended in a medium appropriate for that cell line instead of PBS(-). - Prepare chilled methanol by pouring methanol onto dry ice. Set the temperature of a water bath to 37 °C.
Note: Wear safety glasses to protect eyes when chilled methanol is being prepared and used. - Dip the 15-ml tube into chilled methanol until the cell suspension is completely frozen.
- Transfer the 15-ml tube to the 37 °C water bath, and incubate until the cell suspension is completely thawed.
- Repeat steps C6 and C7 thrice (four cycles in total).
- Centrifuge the 15-ml tube at 400 x g for 5 min at room temperature, and transfer the supernatant to a 1.5-ml or 2-ml cryovial.
- Use the resulting AAV stock immediately for titering or cell infection, or store it at -80 °C until use. Avoid extra freeze-thaw cycles.
Note: The AAV solution prepared as above can be purified using density gradient centrifugation or affinity columns prior to its use for infection, and these procedures have been described in published protocols (Khan et al., 2011; Howes and Schofield, 2015). Although purification of the AAV solution is critical for in vivo use, however, gene targeting in cell lines in vitro can be achieved by infecting cells with this AAV solution without further purification.
- After 72 h of transfection, scrape HEK293(T) cells from the 10-cm culture dish by extensive pipetting, and transfer the cells to a 15-ml conical tube with their supernatant.
- (Optional) AAV titering based on copy number estimation
Although we titered AAV-based targeting vectors when we assessed the gene targeting efficiencies of individual vectors (Karnan et al., 2012; Konishi et al., 2012; Damdindorj et al., 2014; Karnan et al., 2016), titering is optional when the aim of the experiment is only to obtain gene-targeted cell clones.- Perform a serial 10-fold dilution of the maxi-prep plasmid DNA from which AAV particles were produced, and generate six different concentrations of plasmid solutions (10-1-10-6 ng/μl) for use as standard samples in quantitative PCR (qPCR).
- Calculate the copy number of the plasmid in the diluted plasmid solutions. The average molecular weight of deoxyribonucleotides in single-stranded DNA is 303.7. Thus, the approximate copy number of the plasmid N (μl-1) is estimated as:
N = a x NA x 10-9/(n x 303.7)
Where,
a: represents the concentration of plasmid DNA within the solution (ng/μl),
NA: represents Avogadro’s constant (6.022 x 1023),
n: represents the size of the plasmid in base pairs.
Note: In contrast to the single-stranded nature of the AAV vector genome, plasmid DNA is double-stranded under ordinary conditions. However, plasmid DNA is denatured and the copy number is theoretically doubled following the initial denaturation step in qPCR. Therefore, the calculation above was performed accordingly. - In a 1.5-ml tube, mix the following reagents:
AAV stock (prepared in Procedure C) 2.5 μl
RDD buffer 5 μl
DNase I stock solution 1.25 μl
Distilled water 41.25 μl
Note: RDD buffer and DNase I stock solution are components of the RNase-Free DNase Set. - Incubate the reaction mixture above at room temperature (20-25 °C) for 30 min to degrade residual plasmid DNA.
- Incubate the reaction mixture at 95 °C for 5 min to inactivate DNase I.
- Perform qPCR with triplicate templates, including the AAV solution from step D5, serially diluted standard samples, and distilled water (negative control). Initially, prepare a PCR cocktail by mixing the following solutions:
2x buffer for KOD FX Neo 7.5 μl x n
Primer NeoR-Fwd #1 (100 pmol/μl) 0.1 μl x n
Primer NeoR-Rev #2 (100 pmol/μl) 0.1 μl x n
dNTP (10 mM each) 3 μl x n
KOD FX Neo 0.1 μl x n
SYBR Green I working solution 0.15 μl x n
Distilled water 3.05 μl x n
n: the number of templates plus some extra - Dispense the cocktail into a 96-well plate (14 μl/well), add 1 μl of templates to individual wells, and mix by pipetting.
- Perform qPCR amplification using StepOnePlus with the experimental conditions below. The post-amplification melt curve analysis includes 1 min at 95 °C and 1 min at 55 °C followed by a slow temperature increment to 95 °C.
Step 1: 95 °C 2 min
Step 2 (50 cycles): 95 °C 15 sec
60 °C 15 sec
70 °C 20 sec - Calculate the average value of the triplicate data for each sample. Estimate the copy number of an AAV-based targeting vector by comparing its amplification profile with that of serially diluted standard samples. Note that the AAV solution used for qPCR was diluted 20-fold when it was treated with DNase I in step D3. An example of standard curves generated in qPCRs is shown in Figure 8.
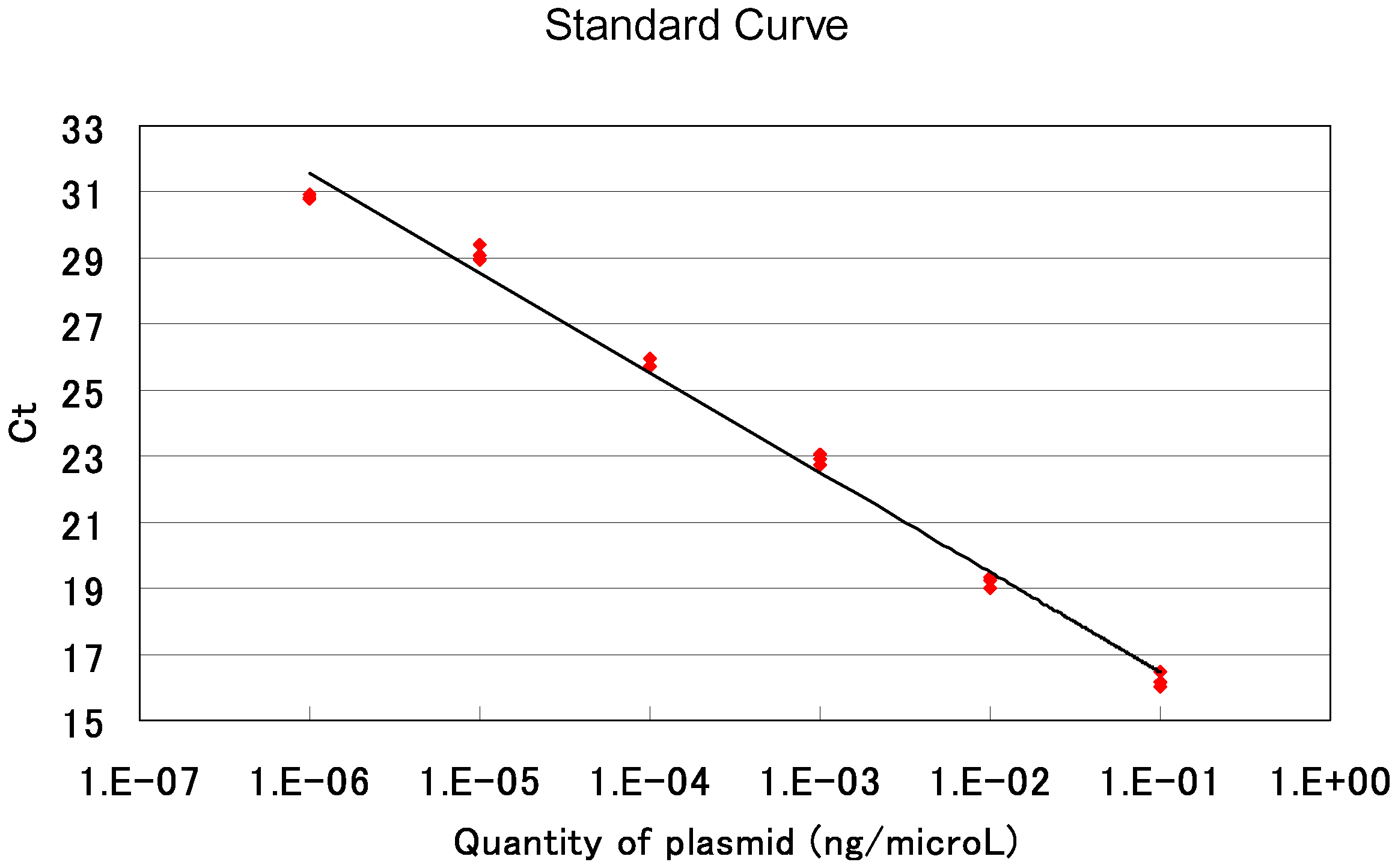
Figure 8. An example of qPCR standard curves. Red dots indicate threshold cycles in individual samples. Ct: Threshold Cycle.
- Perform a serial 10-fold dilution of the maxi-prep plasmid DNA from which AAV particles were produced, and generate six different concentrations of plasmid solutions (10-1-10-6 ng/μl) for use as standard samples in quantitative PCR (qPCR).
- Cell infection with an AAV-based targeting vector
- Propagate the cell line for gene targeting in a 75-cm2 flask with the appropriate medium (growth medium). Cells should be 30-40% confluent on the following day.
- (Optional) If G1/S arrest of the cell line for gene targeting is readily achievable with a simple procedure, then arrest the cells at the G1/S boundary prior to AAV infection, as this was reported to enhance the efficiency of AAV-mediated gene targeting (Trobridge et al., 2005). For instance, MCF-10A cells should be incubated with epidermal growth factor (EGF)-free medium for 6-8 h prior to AAV infection, and the medium should be replaced with EGF-containing medium (growth medium) at the time of infection.
- The day after cell propagation, thaw the AAV stock stored at -80 °C in a 37 °C water bath.
- Aspirate the medium from the 75-cm2 flask, and pour (5-x) ml of growth medium onto the cells, where x (ml) represents a predetermined volume of the AAV stock.
Note: The volume of the AAV stock used is determined by the multiplicity of infection (MOI) that is used to infect the cells. We generally infect cells with AAV-based targeting vectors at a MOI of 1 x 104 (Karnan et al., 2012; Konishi et al., 2012; Damdindorj et al., 2014; Karnan et al., 2016); however, a range of MOI can be applied depending on the experiment (Russell and Hirata, 1998; Hirata and Russell, 2000; Miller et al., 2003; Porteus et al., 2003). If the aim of the experiment is only to obtain gene-targeted cell clones, use the entire (1.0 ml) AAV stock for the 75-cm2 flask. - Add x ml of the AAV stock to the flask, and mix by gentle rocking. Incubate the cells in the CO2 incubator overnight.
- On the next day (16-24 h after infection), add 7 ml of growth medium into the 75-cm2 flask, and incubate the cells in the CO2 incubator for two more days.
- AAV infection is considered complete two days later. The cells can now be used for assays or experiments, although G418 selection is subsequently performed in many cases to eliminate uninfected cells. For single cell cloning with the infected cells, proceed to Procedures F and G.
- Propagate the cell line for gene targeting in a 75-cm2 flask with the appropriate medium (growth medium). Cells should be 30-40% confluent on the following day.
- G418 selection in 96-well tissue culture plates
- Aspirate the AAV-containing medium from the flask, wash the cells with PBS(-) once, and pipette 2 ml of trypsin into the flask. Incubation time with trypsin in the CO2 incubator varies depending on the cell lines employed.
- Swirl the flask and examine it under a microscope to confirm that the cells are dissociated. Then, add the appropriate volume of growth medium to the flask, pipette to further dissociate the cells, and transfer the cell suspension into a 15-ml (or 50-ml) conical tube.
- Centrifuge the cells at 400 x g for 5 min, discard the supernatant, and resuspend the cells in 14 ml of growth medium containing the appropriate concentration of G418.
Note: In this protocol, G418 selection is initiated when cells are seeded into 96-well tissue culture plates as this expedites the selection of cells. However, if the cell line being infected is sensitive to sparse seeding, it is advisable to begin G418 selection 1-3 days after seeding when the cells have attached to the wells. This may increase the number of colonies obtained following G418 selection. - Distribute the cells into seven 96-well plates. First, pour 6 ml of cell suspension to a sterilized pipetting reservoir, and then add 52 ml of growth medium containing G418.
- Mix the cell suspension in the reservoir by pipetting, and inoculate the cells in three 96-well plates (200 μl/well) using a 12-channel pipettor.
- Pour 4 ml of cell suspension to the reservoir, and add 35 ml of growth medium containing G418.
- Mix the cell suspension in the reservoir by pipetting, and inoculate the cells in two 96-well plates (200 μl/well) using a 12-channel pipettor.
- Repeat steps F6 and F7 using the remaining (4 ml) cell suspension.
- Place the 96-well plates in the CO2 incubator, and maintain the cells until completion of G418 selection and detection of colonies by microscopy (usually 7-14 days after seeding).
Note: If colonies in the 96-well plates are not clearly visible because of dying cells or dead cells floating in the medium, the visibility can be improved by changing the medium. - Mark wells containing G418-resistant colonies with a permanent marker. Wells that apparently contain multiple colonies should be marked with a different color.
- Incubate the 96-well plates in the CO2 incubator for another 7-14 days until cells in the marked wells reach an average of 60-80% confluence.
- Aspirate the AAV-containing medium from the flask, wash the cells with PBS(-) once, and pipette 2 ml of trypsin into the flask. Incubation time with trypsin in the CO2 incubator varies depending on the cell lines employed.
- PCR-based screening of cell clones
This section describes a PCR-based screening of pooled samples that are generated by manual one-by-one cell pooling. If the number of colonies formed after G418 selection is relatively small, cell pooling can be omitted and the screening of colonies can be performed on a one-by-one basis (Kohli et al., 2004; Rago et al., 2007; Howes and Schofield, 2011). Systematic multi-round cell pooling that we described previously (Konishi et al., 2007b) is usually unnecessary when the constructed targeting vector is based on pAAV-2Aneo (v2).- Decant or aspirate the medium in the 96-well plates, and wash the cells once with PBS(-).
- Trypsinize the marked cells with trypsin (20 μl/well).
- After cells are dissociated, gently pipette the cell suspension up and down a few times, add growth medium (80 μl/well), and gently pipette the cells again.
- Pipette the cell suspension from 8-12 wells (50 μl/well) into a single 1.5-ml tube. Repeat this for all marked wells of the 96-well plates.
Note: Use a new pipette tip for each cell transfer to prevent cross-contamination of cells. - Add growth medium to the marked wells (150 μl/well), and place the 96-well plates back in the CO2 incubator.
Note: Use a serum-containing medium to neutralize trypsin in this step. If the use of serum is not optimal for the cell line being engineered, replace the serum-containing medium with the appropriate growth medium after the cells have attached to the wells. - Extract genomic DNA from the cell samples pooled in 1.5-ml tubes using the PureLink Genomic DNA Mini Kit.
- Perform screening PCR to identify pooled samples containing gene-targeted cell clones using KOD FX Neo and the genomic DNA extracted in step G6. The PCR-amplified region should encompass one of the homology arms: i.e., one of the PCR primers should correspond to the region unique to the targeting vector, and the other to the region outside of the homology arm (Figures 1 and 5).
- Separate the PCR products by agarose gel electrophoresis, and identify pooled samples yielding a PCR product of the expected size.
- After the cells in the 96-well plates corresponding to the PCR positive hits grow to at least 50 % confluence, trypsinize and transfer the cells to 24-well plates following standard procedures.
- After cells in the 24-well plates grow to at least 50% confluence, aspirate the medium, wash cells once with PBS(-), and treat cells with trypsin (100 μl/well).
- After cells are dissociated, gently pipette the cell suspension up and down a few times, add growth medium (900 μl/well), and gently pipette the cells again.
- Transfer approximately half of the trypsinized cells from each well into a 1.5-ml tube.
- Add growth medium to the 24-well plates up to 1.5 ml/well, and place the plates back in the CO2 incubator.
- Extract genomic DNA from the samples in step G12 using the PureLink Genomic DNA Mini Kit, perform a second screening PCR using KOD FX Neo and the same primer pair as in step G7, and identify PCR-positive samples by agarose gel electrophoresis.
- Verify the PCR-positive products identified in step G14 by direct sequencing using custom-made primers.
- Transfer the PCR-positive cells from the 24-well plates to 6-well plates or tissue culture dishes/flasks. If the positive cells are likely to be of non-single cell origin, perform single cell cloning using a limiting dilution technique, followed by PCR screening of isolated single cell clones using the same primer pair as in steps G7 and G14. Meanwhile, confirm the positivity of PCR encompassing the other homology arm using the genomic DNA obtained in step G14 (primers depicted in Figures 1 and 5). The amplified PCR product of the expected size should be verified by direct sequencing.
Note: To further confirm the integrity of the isolated gene-targeted cell clones, it would be ideal to sequence the whole genome of the clones. However, as an alternative, we usually perform Southern blotting with the isolated gene-targeted clones to confirm the integrity of the targeted genomic locus and the absence of extra copies of the targeting vector that can be randomly integrated into the genome (Karnan et al., 2012; Konishi et al., 2012; Karnan et al., 2016).
- Decant or aspirate the medium in the 96-well plates, and wash the cells once with PBS(-).
Notes
- The promoter-trap module in the targeting vector can be inserted into either an exon or an intron of the endogenous target gene. However, since the neomycin phosphotransferase (NeoR) gene in the targeting vectors is expressed downstream of 2A, the promoter-trap module should be inserted into a coding exon or an intron between the coding exons, and the homology arms should be designed accordingly.
- When the promoter-trap module in a targeting vector is inserted into a coding exon, design the PCR reverse primer to amplify the 5’ homology arm so that a reading frame of the target gene coincides with that shown in Figure 6 or 7. Confirm that neither a stop codon nor a polyadenylation signal will be ectopically introduced at the 3’ end of the 5’ homology arm.
- The sum of the lengths of 5’ and 3’ homology arms should not exceed 3.0 kb because of the size restriction of the AAV vector.
- In amplifying homology arms by PCR to construct a targeting vector, genomic DNA used as a template should ideally be prepared from a cell line used for gene targeting in later steps. The use of a distinct cell line as PCR template potentially results in lower homology between homology arms and the host genome because of polymorphisms within the homologous region. Low homology in the homology arms of a targeting vector results in low gene targeting efficiency. However, to introduce a defined point mutation, deletion, or insertion within a homology arm for targeted knock-in (e.g., Konishi et al., 2007a; Wilsker et al., 2008; Gustin et al., 2009; Konishi et al., 2011), employ a cell line harboring the same genetic alteration with that being knocked-in. Alternatively, perform site-directed mutagenesis after the homology arm is cloned into a vector to introduce the desired alteration.
- Although we have listed here the commercially available reagents, kits, and equipment regularly used in our laboratory, most of them can be replaced with equivalent products from other manufacturers.
Recipes
- Ampicillin working solution
- Dissolve 1 g of ampicillin sodium in 20 ml distilled water (50 mg/ml).
- Sterilize the solution using a 0.22 µm filter.
- Aliquot the sterilized solution into 1.5-ml tubes (1 ml/tube).
- Store the aliquots at -20 °C.
- SYBR Green I working solution
- Aliquot SYBR Green I to 1.5-ml tubes (1 μl/tube). Immediately wrap the tubes with aluminum foil to protect from light, and label them as ‘80,000x’.
- Store the ‘80,000x’ tubes at -20 °C (except for a tube which will be processed as below). Use non-clear boxes for storage.
- Label 40 of 1.5-ml tubes as ‘2,000x’.
- Dilute 1 μl of SYBR Green I in a ‘80,000x’ tube with 39 μl of DMSO, and aliquot the resultant 40 μl solution to 40 of ‘2,000x’ tubes (again 1μl/tube).
- Store the ‘2,000x’ tubes at -20 °C. Use non-clear boxes for storage.
- When preparing a qPCR cocktail, thaw a ‘2,000x’ tube and dilute the content with 19 μl of distilled water. This results in the generation of 100x SYBR Green I working solution (20 μl). Discard excess 100x working solution in a thawed tube after qPCR setup.
- PBS(-)
PBS(-) is prepared according to standard procedures.- First, the ingredients below are dissolved in distilled water to produce 10x PBS(-).
160.2 g NaCl
4.0 g KCl
58.0 g Na2HPO4.12H2O
4.0 g KH2PO4
Distilled water to 2 L - Then, 50 ml of 10x PBS(-) is diluted with 450 ml of distilled water in a 500-ml bottle and sterilized by autoclave.
- First, the ingredients below are dissolved in distilled water to produce 10x PBS(-).
Acknowledgments
This protocol was adapted from our previous work, Karnan et al. (2016). This work was supported by Grants-in-Aid for Scientific Research (KAKENHI) from the Japan Society for the Promotion of Science (JSPS; 25460395 to S. K., 15K19561 to A. O., and 25640107 to H. K.), and Takeda Science Foundation (to H. K.).
References
- Damdindorj, L., Karnan, S., Ota, A., Hossain, E., Konishi, Y., Hosokawa, Y. and Konishi, H. (2014). A comparative analysis of constitutive promoters located in adeno-associated viral vectors. PLoS One 9(8): e106472.
- Gustin, J. P., Karakas, B., Weiss, M. B., Abukhdeir, A. M., Lauring, J., Garay, J. P., Cosgrove, D., Tamaki, A., Konishi, H., Konishi, Y., Mohseni, M., Wang, G., Rosen, D. M., Denmeade, S. R., Higgins, M. J., Vitolo, M. I., Bachman, K. E. and Park, B. H. (2009). Knockin of mutant PIK3CA activates multiple oncogenic pathways. Proc Natl Acad Sci U S A 106(8): 2835-2840.
- Hirata, R. K. and Russell, D. W. (2000). Design and packaging of adeno-associated virus gene targeting vectors. J Virol 74(10): 4612-4620.
- Howes, R. and Schofield, C. (2015). Genome engineering using Adeno-Associated Virus (AAV). Methods Mol Biol 1239, 75-103.
- Karnan, S., Konishi, Y., Ota, A., Takahashi, M., Damdindorj, L., Hosokawa, Y. and Konishi, H. (2012). Simple monitoring of gene targeting efficiency in human somatic cell lines using the PIGA gene. PLoS One 7(10): e47389.
- Karnan, S., Ota, A., Konishi, Y., Wahiduzzaman, M., Hosokawa, Y. and Konishi, H. (2016). Improved methods of AAV-mediated gene targeting for human cell lines using ribosome-skipping 2A peptide. Nucleic Acids Res 44(6): e54.
- Khan, I. F., Hirata, R. K. and Russell, D. W. (2011). AAV-mediated gene targeting methods for human cells. Nat Protoc 6(4): 482-501.
- Kohli, M., Rago, C., Lengauer, C., Kinzler, K. W. and Vogelstein, B. (2004). Facile methods for generating human somatic cell gene knockouts using recombinant adeno-associated viruses. Nucleic Acids Res 32(1): e3.
- Konishi, H., Karakas, B., Abukhdeir, A. M., Lauring, J., Gustin, J. P., Garay, J. P., Konishi, Y., Gallmeier, E., Bachman, K. E. and Park, B. H. (2007a). Knock-in of mutant K-ras in nontumorigenic human epithelial cells as a new model for studying K-ras mediated transformation. Cancer Res 67(18): 8460-8467.
- Konishi, H., Lauring, J., Garay, J. P., Karakas, B., Abukhdeir, A. M., Gustin, J. P., Konishi, Y. and Park, B. H. (2007b). A PCR-based high-throughput screen with multiround sample pooling: application to somatic cell gene targeting. Nat Protoc 2(11): 2865-2874.
- Konishi, H., Mohseni, M., Tamaki, A., Garay, J. P., Croessmann, S., Karnan, S., Ota, A., Wong, H. Y., Konishi, Y., Karakas, B., Tahir, K., Abukhdeir, A. M., Gustin, J. P., Cidado, J., Wang, G. M., Cosgrove, D., Cochran, R., Jelovac, D., Higgins, M. J., Arena, S., Hawkins, L., Lauring, J., Gross, A. L., Heaphy, C. M., Hosokawa, Y., Gabrielson, E., Meeker, A. K., Visvanathan, K., Argani, P., Bachman, K. E. and Park, B. H. (2011). Mutation of a single allele of the cancer susceptibility gene BRCA1 leads to genomic instability in human breast epithelial cells. Proc Natl Acad Sci U S A 108(43): 17773-17778.
- Konishi, Y., Karnan, S., Takahashi, M., Ota, A., Damdindorj, L., Hosokawa, Y. and Konishi, H. (2012). A system for the measurement of gene targeting efficiency in human cell lines using an antibiotic resistance-GFP fusion gene. Biotechniques 53(3): 141-152.
- Miller, D. G., Petek, L. M. and Russell, D. W. (2003). Human gene targeting by adeno-associated virus vectors is enhanced by DNA double-strand breaks. Mol Cell Biol 23(10): 3550-3557.
- Porteus, M. H., Cathomen, T., Weitzman, M. D. and Baltimore, D. (2003). Efficient gene targeting mediated by adeno-associated virus and DNA double-strand breaks. Mol Cell Biol 23(10): 3558-3565.
- Rago, C., Vogelstein, B. and Bunz, F. (2007). Genetic knockouts and knockins in human somatic cells. Nat Protoc 2(11): 2734-2746.
- Russell, D. W. and Hirata, R. K. (1998). Human gene targeting by viral vectors. Nat Genet 18(4): 325-330.
- Trobridge, G., Hirata, R. K. and Russell, D. W. (2005). Gene targeting by adeno-associated virus vectors is cell-cycle dependent. Hum Gene Ther 16(4): 522-526.
- Wilsker, D., Petermann, E., Helleday, T. and Bunz, F. (2008). Essential function of Chk1 can be uncoupled from DNA damage checkpoint and replication control. Proc Natl Acad Sci U S A 105(52): 20752-20757.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Karnan, S., Ota, A., Konishi, Y., Wahiduzzaman, M., Tsuzuki, S., Hosokawa, Y. and Konishi, H. (2016). Efficient AAV-mediated Gene Targeting Using 2A-based Promoter-trap System. Bio-protocol 6(24): e2058. DOI: 10.21769/BioProtoc.2058.
Category
Molecular Biology > DNA > DNA cloning
Molecular Biology > DNA > DNA recombination
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.